
Abstract
Bupivacaine is frequently used for conducting regional anesthesia. When accidentally injected or excessively absorbed into circulation, bupivacaine can induce severe arrhythmia and potentially lead to cardiac arrest. The specific mechanisms underlying this cardiotoxicity, however, remain to be clarified. We transfected HEK-293 cells to express the small conductance calcium-activated potassium type-2 channel (SK2), and used a whole-cell patch clamp method in order to explore how bupivacaine affected these channels. We subsequently used SK2 knockout mice to explore the relevance of SK2 channels in bupivacaine-induced cardiotoxicity in isolating mouse hearts, mounting them on a Langendorff apparatus, and perfusing them with bupivacaine. Using this system, arrhythmia, asystole, and cardiac functions were monitored. We observed dose-dependent inhibition of SK2 channels by bupivacaine: half-maximal inhibitory concentration (IC50) value = 18.6 μM (95% CI 10.8–32.1). When SK2 knockout (SK2 −/−) or wild-type (WT) mice were perfused with Krebs-Henseleit buffer (KHB), we did not observe any instances of arrhythmia. When SK2 −/− mice or WT were perfused with KHB containing bupivacaine (40 μM), the time to arrhythmia (Tarrhythmia) and time to asystole (Tasystole) were both significantly longer in SK2 −/− mice relative to WT mice (P < 0.001). Similarly, SK2 −/− mice exhibited a significantly longer time to 25%, 50%, and 75% reductions in heart rate (HR) and rate-pressure product (RPP) relative to WT mice following bupivacaine perfusion (P < 0.001). These results reveal that bupivacaine was able to mediate a dose-dependent inhibition of SK2 channels in HEK-293 cells, and deletion of SK2 channels can delay bupivacaine-induced cardiotoxicity in isolated mouse hearts.
Introduction
Regional anesthesia achieved through local administration of local anesthetics (LAs) is commonly employed for obstetric or orthopedic surgical operations, and for achieving postoperative analgesia. 1,2 Bupivacaine is an amide LA that is commonly used in the clinic owing to its desirable analgesic properties and extended duration of efficacy. 3 Typically, bupivacaine is safe, but in some cases the accidental administration or excess absorption of this LA into circulation can lead to severe arrhythmia and cardiac arrest, leading to poor patient outcomes. 4 –6 As such, anesthesiologists need to be aware of the optimal strategies for both preventing and treating bupivacaine-induced cardiotoxicity.
As the mechanisms governing bupivacaine-induced cardiotoxicity are complex, they remain to be fully elucidated. 4 This toxicity is known to be associated with the ability of bupivacaine to substantially inhibit ion channel and receptor activity on cardiomyocytes. 4 In particular, research has suggested that this toxicity is a secondary consequence of the inhibition of sodium, 7 L-calcium, 8 and transient outward potassium current channels. The sodium current is the primary current important for mediating depolarization during action potential formation (phase 0), thereby controlling cardiomyocyte excitability and conductivity. 9 As bupivacaine can interfere with this depolarization in the context of cardiomyocyte action potentials through a mechanism linked with impaired Na+ influx, this can result in subsequent arrhythmias and potentially cardiac arrest. 10 These findings led to the conclusion that inhibition of sodium channels is the primary mechanism whereby bupivacaine induces cardiotoxicity. 7,11 However, several additional ion channels have more recently been found to play a role in controlling the excitation and conduction processes of cardiomyocytes. 12,13 As such, it is important that the possible role of these receptors in bupivacaine-induced cardiotoxicity be investigated.
Calcium-activated potassium channels are triggered in response to changes in levels of calcium within cells, and serve as key regulators of normal cardiomyocyte functionality. 13,14 There are three categories of calcium-activated potassium channels in humans: large, medium, and small conductance channels. Small conductance calcium-activated potassium type 2 channel (SK2) is an important potassium channel that is involved in the hyperpolarization that occurs following an action potential. 12,13,15 These SK2 channels are present in both the nervous and circulatory system, with distribution on atrial cells, 16 ventricular cells, 16 and Purkinje cells, 12 playing vital roles in regulating cardiac conduction. Indeed, these SK2 channel have recently been shown to play a direct role in atrial fibrillation in humans, 12 with SK2 channel upregulation contributing to heart failure. 17,18 Given their key role in the context of action potential propagation, the dysfunction of SK2 channels has the potential to induce atrial or ventricular arrhythmia. 19 Bupivacaine-induced cardiotoxicity is known to occur due to disruption in the activation of cardiac ion channels, 20 but the effects of bupivacaine on SK2 channels is yet to be reported. We therefore hypothesized that bupivacaine may directly suppress SK2 currents, thereby contributing to the cardiotoxicity associated with this LA agent.
In this report, we utilized HEK-293 cells expressing SK2 channels for whole-cell patch clamp studies, and we further utilized isolated hearts from SK2-knockout mice in order to explore the role of SK2 channels in bupivacaine-induced cardiotoxicity.
Method
Chemicals
Human embryonic kidney 293 cells (HEK293 cells) were from the Institute of Life Sciences of the Chinese Academy of Sciences. Additionally, the following were purchased: Dulbecco’s modified Eagle’s medium (DMEM) from Hyclone (USA); fetal bovine serum (FBS), penicillin/streptomycin, and 0.25% trypsin from Gibco (USA); pCDNA3/rSK Ca2+ plasmids from OriGene (USA); Lipofectamine 2000 from Invitrogen (USA); and bupivacaine from Sigma Aldrich (USA). The pipette solution was composed of the following, in mM: MgCl2 1.15, Potassium gluconate 144, and CaCl2 1.0, pH was maintained at 7.3 with KOH.
Cell culture and transfection
HEK293 Cells were grown using DMEM containing 10% FBS at 37°C in a 5% CO2 incubator with penicillin/streptomycin, and 0.25% trypsin was used for detecting cells. Prior to transfection cells were plated 2 × 105 cells/cm2, and cells were grown to 85% confluency at which time cells were transfected with the pCDNA3/rSK Ca2 plasmids using Lipofectamine 2000 based on provided directions. This allowed us to generate HEK-293 cells stably expressing SK2 channels (SK2 cells), as described previously. 21 Prior to patch clamp studies, SK2 cells were played in a glass cover for 24 h.
SK2 current recordings
A whole-cell patch clamp approach was used as described previously. 22 The coverslip containing the SK2 cells was placed into an inverted Olympus microscope (IX70, Japan) on the cell chamber. A superfusion system was then used for the assay, with appropriate solutions added to reservoirs. The system used was a DAD-VC system with a Micromanifold made up of eight polyamide-coated quarts tubes (100 um ID). This manifold allows for the simultaneous flow of up to eight different solutions into a single <1 uL, and can be readily maneuvered around cells to accurately direct it at target cells.
An EPC-10 amplifier was used for whole-cell patch clamping. Briefly, a glass electrode with a 1.2 mm outer diameter was pulled with a microelectrode puller, yielding a 1.5–3.0 MΩ resistance after the pipette solution was added. We then picked SK2 cells with smooth cell membranes using microscopic guidance and recorded currents for these cells. After achieving a giga-seal, negative pressure was used to disrupt SK2 cell membrane, and the Pulse 8.0 software (HEKA, Germany) was used for voltage stimulation and data recording, with all experiments being conducted at 36°C. The SK2 currents stimulation program was as follows: a holding potential was maintained at −80 mV, the step clamp voltage was from −120 mV to +40 mV with a step of 10 mV and a 200 ms duration. Cells were able to produce stable currents at 0 mV; therefore, currents at 0 mV were used for comparisons in the following experiments.
Preparation of Langendorff-isolated heart
Age-, sex- and weight-matched adult SK2 knockout and WT C57BL/6J mice (8–10 weeks old) were used for this study. The SK2 −/− mice were generated, validated, and bred onto a C57BL/6J background at the Nanjing Model Institute of Zoology. All experiments were conducted in the laboratory of Wenzhou Medical University with approval from the Animal Care and Use Committee of Wenzhou Medical University. Animal care and treatments were carried out in accordance with the guidelines of the National Institutes of Health.
Mice were intraperitoneally administered 80 mg/kg ketamine hydrochloride and 12 mg/kg xylazine to achieve anesthesia, after which they were injected with 500U/kg heparin to prevent intracoronary microthrombi formation. We then rapidly excised the hearts from these animals and subjected them to retrograde perfusion with modified Krebs-Henseleit buffer (KHB; NaCl: 118 mmol/L, KCl: 4.7 mmol/L, pH: 7.40 ± 0.05) at 37°C and 80 mmHg. A latex balloon in the left ventricle was used for monitoring left ventricular pressure, with saline being intermittently injected into this balloon so as to keep left ventricular end-diastolic pressure between 4 mmHg and 6 mmHg. Electrocardiograph (ECG) electrodes were consistently placed in a “lead Ⅱ” position. The PowerLab biological signal processing and analysis system and the Chart 5.5.6 biological signal recording software were used for all data collection. The experimental protocol was initiated following 25 minutes of KHB perfusion, at which time steady-state conditions had been achieved.
Assessment of cardiotoxicity
Cardiotoxicity was first assessed via perfusing normal modified KHB through the isolated hearts of WT and SK2 −/− mice (n = 8 per group) (Experimental Protocol was illustrated in Figure 1). In addition, to test the effects of bupivacaine on cardiotoxicity, WT and SK2 −/− mice were perfused with KHB containing 40 uM bupivacaine (0.5%, Sigma-Aldrich Co., St. Louis, MO, P code: B5274-5G) (n = 8 per group). The times corresponding to the following parameters were measured for all groups: the time to first ventricular premature beat following bupivacaine administration (Tarrhythmia); the time to asystole following bupivacaine administration (Tasystole); and the time to 25%, 50%, and 75% reductions in heart rate (HR) and rate-pressure product (RPP = HR × (systolic pressure − diastolic pressure)) relative to baseline following bupivacaine administration. Baseline hemodynamic parameters were established prior to bupivacaine infusion.

Experimental protocol. WT: wild type mice; SK2 −/−: SK2 knockout mice; B: bupivacaine; Tasystole: time to asystole.
Statistical analyses
All statistical analyses were conducted using SPSS (19.0, IL, USA). Shapiro-Wilk tests were used to assess the normality of measured data. The dose-dependent ability of bupivacaine to inhibit SK2 currents was assessed via non-linear fitting of the data using GraphPad Prism 5.0 (CA, USA): Y = Bottom + (Top-Bottom)/(1 + 10^((LogIC50-X)*HillSlope)). Continuous data were given as means ± standard deviation. Weights, Tarrhythmia and Tasystole were assessed via one-way ANOVAs with LSD tests used for pairwise comparisons of significant results. Cardiac function parameters were compared via repeated-measured ANOVAs, with Scheffe tests being used to facilitate multiple comparisons. P < 0.05 was the significance threshold.
Results
Bupivacaine inhibits SK2 currents in a dose-dependent fashion
We first used HEK-293 cells which were transfected to express SK2 channels in order to assess the ability of bupivacaine to inhibit SK2 currents. We found that increasing bupivacaine concentrations were associated with a reduction in SK2 current. We were able to fit this dose-response data for bupivacaine inhibition of SK2 currents (at 0 mV) using a non-linear model, yielding a bupivacaine half-maximal inhibitory concentration (IC50) value of 18.6 μM (95% CI 10.8–32.1) (Figure 2).

Bupivacaine inhibits SK2 currents in a dose-dependent manner. The ability of bupivacaine to inhibit SK2 currents (0 mV) in a dose-dependent fashion was assessed via non-linear fitting to yield the IC50 value shown. The error bar indicates the standard deviation from the mean value. The equation was Y = Bottom + (Top-Bottom)/(1 + 10^((LogIC50-X)*HillSlope)). A total of 1 μM free calcium was present within the pipette.
Baseline parameters, Tarrhythmia and Tasystole for mouse studies
There were no differences in baseline weight (23.2 ± 3.1, 22.5 ± 2.5, 23.4 ± 2.3, and 23.3 ± 3.4 g), HR (394.1 ± 7.4, 396.6 ± 7.8, 401.5 ± 11.7 and 396.5 ± 9.5 beats/s) or RPP (3741.0 ± 1081.0, 38322.9 ± 1771.1, 37673.8 ± 3610.2, and 37577.0 ± 1831.3 mm Hg·beats/s) in the WT, SK2−/−, WT + B, or SK2−/− + B groups, respectively. Hearts in the SK2−/− + B group had significantly longer Tarrhythmia and Tasystole values than did hearts in the WT + B group (both P < 0.001). There was no occurrence of arrhythmia in the WT or SK2−/− group (Table 1).
Times to defined HR, RPP, and ECG endpoints after bupivacaine perfusion.a

WT: wild type mice; SK2 −/−: SK2 knockout mice; B: bupivacaine. Tarrhythmia: the time to first ventricular premature beat after perfused with KHB containing 40 uM bupivacaine; Tasystole: the time to asystole after perfused with KHB containing 40 uM bupivacaine; HR: heart rate; RPP: rate-pressure product.
a Data are given as the mean ± standard deviation.
Cardiac function variables
Following bupivacaine infusion initiation, the hearts in the SK2 −/− + B group exhibited a significantly longer time to 25%, 50%, and 75% reduction in HR and RPP relative to those in the WT + B group (Figures 3 and 4). Within 1–7 minutes of bupivacaine perfusion, the HR and RPP values in the SK2 −/− + B group had significantly increased than the values in WT + B group (Figures 3 and 4).
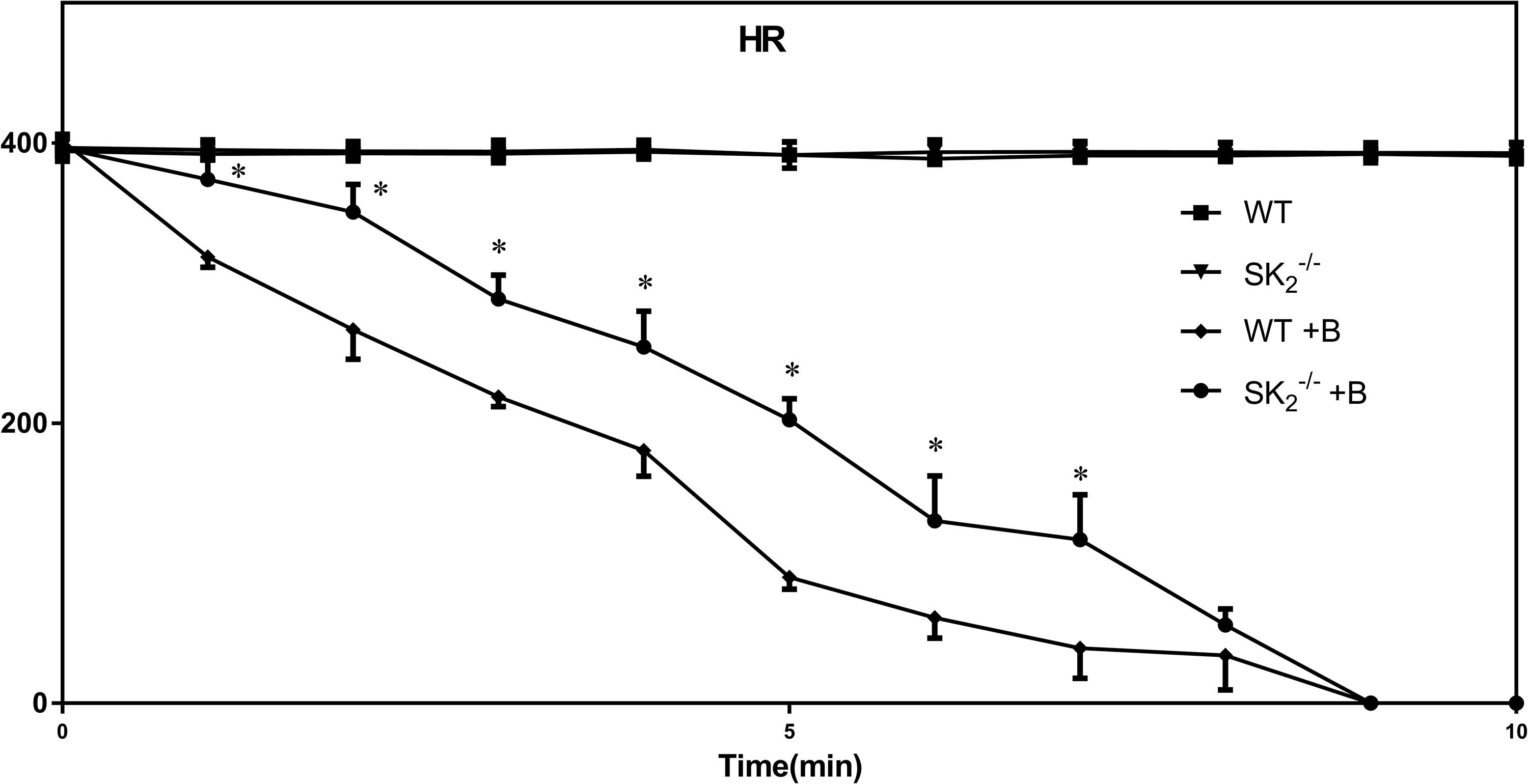
Changes in HR (beats/s) among the four study groups (n = 8 for each group). HRs (Heart Rate) for the four treatment groups are shown. Data are means ± standard deviation. The error bar on each data point indicates the standard deviation from the mean value. *P < 0.05, between WT + B and SK2 −/− + B groups. WT: wild type mice; SK2 −/−: SK2 knockout mice; B: bupivacaine.

Changes in RPP (mm Hg·beats/s) among the four study groups (n = 8 for each group). RPPs (Rate-pressure product) for the four treatment groups are shown. Data are means ± standard deviation. The error bar on each data point indicates the standard deviation from the mean value. *P < 0.05, between WT + B and SK2 −/− + B groups. WT: wild type mice; SK2 −/−: SK2 knockout mice; B: bupivacaine.
Discussion
The results of this study revealed that bupivacaine was capable of dose-dependent inhibition of SK2 currents, with an IC50 value of 18.6 µM. We further found that deleting SK2 channels was associated with a significant increase in time to first arrhythmia, time to asystole, and time to 25%, 50%, and 75% reductions in HR and RPP following cardiac bupivacaine infusion.
The isolated cardiac perfusion model has certain advantages. First, it can exclude the systematic effect of bupivacaine on our observations of bupivacaine cardiotoxicity. In addition, the interference factor in this model is small, which can reduce the total requirement of knockout mice. The bupivacaine concentration (40 µM) used in the present study was derived from our previous work, 23 at which all isolated hearts stop beating. Due to ventricular premature was very easy to identify on the electrocardiogram, so we defined it as arrhythmia in the study.
There are several different types of ion channels that play essential roles in regulating cardiac function, including sodium channels, 7 L-calcium channels, 24 transient outward potassium channels, 25 and beta adrenergic receptors. 26 Abnormal functioning of these channels or receptors can disrupt normal cardiac electrophysiology and systolic function, and its abnormalities can affect the normal functioning of the heart. Sodium channels are particularly important for action potential depolarization, whereas L-calcium channels are important for the repolarization plateau, and transient outward potassium channel currents are vital for normal repolarization. The disrupted function of these three types of ion channels has previously been proposed as the primary mechanism underlying bupivacaine-induced cardiotoxicity. 10,27,28 However, there are many other ion channels within the cardiac tissue, raising the possibility that other pathways are also involved in this toxicity.
Very few researchers have assessed the impact of bupivacaine on calcium-activated potassium channels. Martín et al. 29 assessed the ability of bupivacaine to inhibit large conductance calcium-activated potassium channels (BK) using human umbilical artery smooth muscle cells. In so doing he observed dose-dependent BK inhibition mediated by bupivacaine. However, no previous reports have explored the ability of bupivacaine to affect SK2 channel activity. In the present study, we found that bupivacaine inhibited currents through SK2 channels on HEK293 cells, with an IC50 value of 18.6 μM. This suggests that SK2 currents are sensitive to bupivacaine. In clinical instances where bupivacaine accidentally enters the bloodstream, circulating levels can readily exceed 10 μM. To further verify the role of SK2 channels in bupivacaine-induced cardiotoxicity, we therefore next conducted in vivo studies using mice in which this ion channel had been knocked out.
Following knockout of certain genes, compensatory pathways are often engaged, leading to the lack of any apparent phenotype under baseline conditions following gene knockout. 30,31 Consistent with this, SK2 knockout mice did not exhibit arrhythmia at baseline. This may be because a loss of SK2 channel expression in the cardiomyocytes may have led to the induction of compensatory mechanisms that achieved normal cardiomyocyte homeostasis, although the specific pathways involved remain uncertain.
We hypothesized that SK2 channel knockout may help to reduced bupivacaine-induced cardiotoxicity as these channels are targeted by bupivacaine. Consistent with this hypothesis, SK2 knockout mice exhibited significantly longer times to arrhythmia and cardiac arrest following bupivacaine induction than did WT mice. This thus revealed that SK2 gene knockout delayed the onset of bupivacaine-induced cardiotoxicity in isolated mouse hearts.
SK2 currents can be blocked by apamin, with Chang et al. 32 having shown that apamin induced early afterdepolarizations, premature ventricular beats, and torsades de pointes ventricular arrhythmia from area failing rabbit ventricles exhibiting secondary. In addition, SK2 gene variants are linked to lethal ventricular arrhythmias in patients suffering from underlying heart diseases. 18 This suggests that inhibiting SK2 current can alter normal cardiac conductivity. Our present results further reveal that bupivacaine is capable of blocking SK2 currents, which likely contributes to bupivacaine-induced cardiotoxicity. While SK2-knockout mice did eventually suffer from cardiac arrest, there was a significant delay in arrhythmia development relative to WT mice, suggesting that the knock out of this gene improved bupivacaine resistance in mice isolated hearts.
Limitations
In this report we utilized HEK-293 cells expressing SK2 channels for whole-cell patch clamp experiments. Such an approach was limited by the fact that this expression was not physiological, and so may not be representative of normal human or mouse cardiomyocytes. Furthermore, we have not explored potential compensatory mechanisms which may be active in mice following knockout of the SK2 gene.
Conclusion
Bupivacaine was able to mediate a dose-dependent inhibition of SK2 channels in HEK-293 cells. Knockout of SK2 channel was associated with a delay in the ability of bupivacaine to induce cardiotoxicity in mouse isolated hearts. Our team will try to further verify our results in vivo.
Footnotes
Abbreviations
Acknowledgment
The authors would like to thank Thomas J Papadimos of University of Toledo College of Medicine and Life Sciences and Yun Xia of the Ohio State University Medical Center for revising the manuscript.
Author contributions
HC participated in conduct of the study, data collection, manuscript preparation, and data analysis; FX was responsible for study design, conduct of the study, data analysis, and manuscript preparation; XC and YC were responsible for study design, conduct of the study, data analysis, and study data collection; and ZJ was responsible for study design, conduct of the study, and manuscript preparation.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by the Natural Science Foundation of Zhejiang Province, China [Grant Number: LQ18H090006] and Wenzhou Municipal Scientific and Technological Program Projects [Grant Number: Y20190410].