
Abstract
Bupivacaine is frequently used for regional anesthesia and postoperative analgesia. However, an inadvertent intravenous injection can cause severe cardiotoxicity, manifesting as arrhythmia, hypotension, and even cardiac asystole. The mechanism of bupivacaine-mediated cardiotoxicity remains unclear. SK2 knockout mice (SK) and wild-type mice (WT) were divided into four groups, with 12 mice per group. We determined the difference in bupivacaine cardiotoxicity between SK2 knockout and WT mice by measuring the time to the first arrhythmia (Tarrhythmia) and the time to asystole (Tasystole). Secondary indicators of cardiotoxicity were the time from the beginning of bupivacaine infusion to 20% prolongation of the QT interval (TQT) and the time to 20% widening of the QRS complex (TQRS). Tarrhythmia and Tasystole were significantly longer in the SK-bupi group than in the WT-bupi group (both P < 0.05). TQT and TQRS were longer in the SK-bupi group than in the WT-bupi group (all P < 0.05). The time to 25%, 50%, and 75% reduction in HR in the SK-bupi group was significantly longer than in the WT-bupi group (all P < 0.05). Knocking out the SK2 channel can reduce bupivacaine-induced cardiotoxicity in the mouse.
Introduction
Amide local anesthetics, such as bupivacaine, are frequently used for regional anesthesia and postoperative analgesia. They have the advantages of fast onset time and long action time. 1 However, bupivacaine can cause severe cardiotoxicity, manifesting as arrhythmia, hypotension, and even cardiac asystole.2–5 Currently, the consensus is that bupivacaine triggers cardiotoxicity by blocking ion channels in cardiomyocytes, including voltage-gated sodium channels, L-type calcium channels, and transient outward potassium channels.6–8
Type 2 small conductance calcium-activated potassium (SK2) channel is a newly discovered ion channel. 9 SK2 channels are widely distributed in atrial, ventricular myocytes, and Purkinje cells, 10 and are essential for the normal electrophysiological activity of the heart. 11 It has been demonstrated12–14 that the SK2 channel is closely related to atrial fibrillation and pathological arrhythmia. It has been previously shown that a low concentration of bupivacaine inhibits the SK2 current. It was also found that the knockout of the SK2 channel gene could suppress the cardiotoxicity of bupivacaine in the isolated mouse heart. 15 However, this effect was not verified in vivo.
Our primary hypothesis was that SK2 channel gene deletion could also increase the tolerance of mice to bupivacaine. In this study, we observed the cardiotoxicity of bupivacaine between SK2 knockout mice and wild-type mice. The main indicators of cardiotoxicity were the time to the first arrhythmia (Tarrhythmia) and the time to asystole (Tasystole). Secondary indicators included the time from the beginning of bupivacaine infusion to 20% prolongation of the QT interval (TQT) and the time to 20% widening of the QRS complex (TQRS).
Material and methods
Experimental animals
The SK2 knockout mice (C57BL/6J background) and wild-type mice were purchased from the Nanjing Animal Model Company (Nanjing, China). The mice were housed in the animal center of Wenzhou Medical University. The temperature was 26°C, and the humidity was 70%. Mice were used for experiments after 2 weeks of acclimatization. All experiments were approved by the Animal Ethics Committee of Wenzhou Medical University (approval number: wydw2019-0227). All procedures involving the use of animals were conducted in compliance with the guidelines of the National Institute of Health.
Anesthesia and monitoring
Before the experiments, mice were fasted for 12 hours and anesthetized by intraperitoneal injection of chloral hydrate (350 mg/kg). After tracheotomy, animals were mechanically ventilated with pure oxygen and 0.5% sevoflurane. The tidal volume was 6 ml/kg, respiratory rate was 80 times/min, and the inhalation-to-exhalation ratio was 2:3. Subsequently, the right internal jugular vein was cannulated to administer bupivacaine (P code: B5274-5G, Sigma-Aldrich, St. Louis, MO, USA). The central venous pressure was monitored and kept at 5–10 mmHg. At the same time, the left carotid artery was catheterized and connected with the Medlab biological signal acquisition system to measure arterial pressure. Needle electrodes were inserted subcutaneously into both upper limbs and left lower limbs, and ECG was recorded. Fifteen minutes after the operation, carotid blood was drawn for blood gas analysis. Only mice with normal results of blood gas analysis were used in the experiments.
Experimental design
SK2 knockout mice were randomly divided into SK and SK-bupi groups, and wild-type mice were divided into WT and WT-bupi groups; each group contained 12 mice. The SK and WT groups were infused with normal saline at the rate of 2.5 mg/kg/min, while the SK-bupi and WT-bupi groups were infused with 1% bupivacaine at the rate of 2.5 mg/kg/min until cardiac asystole (Figure 1). The time of initiating the infusion of bupivacaine was marked as T0. The following parameters were recorded: (1) The time to arrhythmia (Tarrhythmia) and the time to cardiac asystole (Tasystole). Tarrhythmia was defined as the time from bupivacaine infusion to the emergence of arrhythmia, and Tasystole was defined as the time from bupivacaine infusion to cardiac asystole. Arrhythmia was defined as a dropped beat of the heart. 11 (2) QT interval and QRS wave width. The values were used to calculate the time from the beginning of bupivacaine infusion to 20% prolongation of the QT interval (TQT) and the time to 20% widening of the QRS complex (TQRS). (3) Heart rate (HR) after bupivacaine infusion. (4) Mean arterial pressure (MAP) after bupivacaine infusion. (5) The plasma and myocardial concentration of bupivacaine.

Timeline of the experiment. WT: wild-type mice; SK: SK2 knockout mice; BUPI: bupivacaine; Tasystole: time to asystole.
Determination of bupivacaine concentration
After cardiac asystole, the thoracic cavity was opened, the blood was collected by cardiac puncture with a 1 ml syringe, centrifuged at 4000 rpm for 15 min, and the supernatant and heart samples were collected and frozen at −80°C. 16 The plasma and myocardial concentrations of bupivacaine were determined by liquid chromatography-mass spectrometry (HPLC-MS).
Statistics
The power analysis was based on our preliminary study using the Power Sample Size (PASS 11.0) software program. We compared Tarrhythmia among the various treatment groups. Using a two-tailed type 1 error of 5% and type 2 error of 10% (α = 0.05, β = 0.1), the sample size of nine per group was obtained. To account for this result and limit the use of animals, 12 rats per group were used in this study.
Statistical analyses were performed using the SPSS 19.0 (SPSS Inc., Chicago, IL, USA) software. The values are expressed as mean ± SD. Shapiro-Wilk test was used to determine the normal distribution of the data. The baseline values of hemodynamics and blood gas analysis, Tarrhythmia, Tasystole, TQT, TQRS, and plasma and myocardial concentration of bupivacaine were analyzed by one-way ANOVA, and the LSD test was used for pairwise comparison among groups. The value of non-normal distribution was tested by the Kruskal-Wallis H test. P < 0.05 was considered statistically significant.
Results
There were no significant differences in body weight, MAP, HR, and blood gas analysis results among the four groups (Table 1).
Comparison of body weight, cardiac function parameters, and arterial blood gas basic values of four groups of mice.

WT: infusion of normal saline into WT mice; SK: infusion of bupivacaine in SK2 knockout mice; WT-bupi: infusion of bupivacaine in WT mice; SK-bupi: infusion of bupivacaine in SK2 gene knockout mice; HR: heart rate; MAP: mean arterial pressure. Normally distributed data were expressed as mean ± standard deviation.
Arrhythmia time and cardiac asystole time
Tarrhythmia and Tasystole in the SK-bupi group were significantly longer than those in the WT-bupi group (P < 0.05) (Figure 2(a) and (b)). No instances of arrhythmia and cardiac asystole were detected in the SK and WT groups.
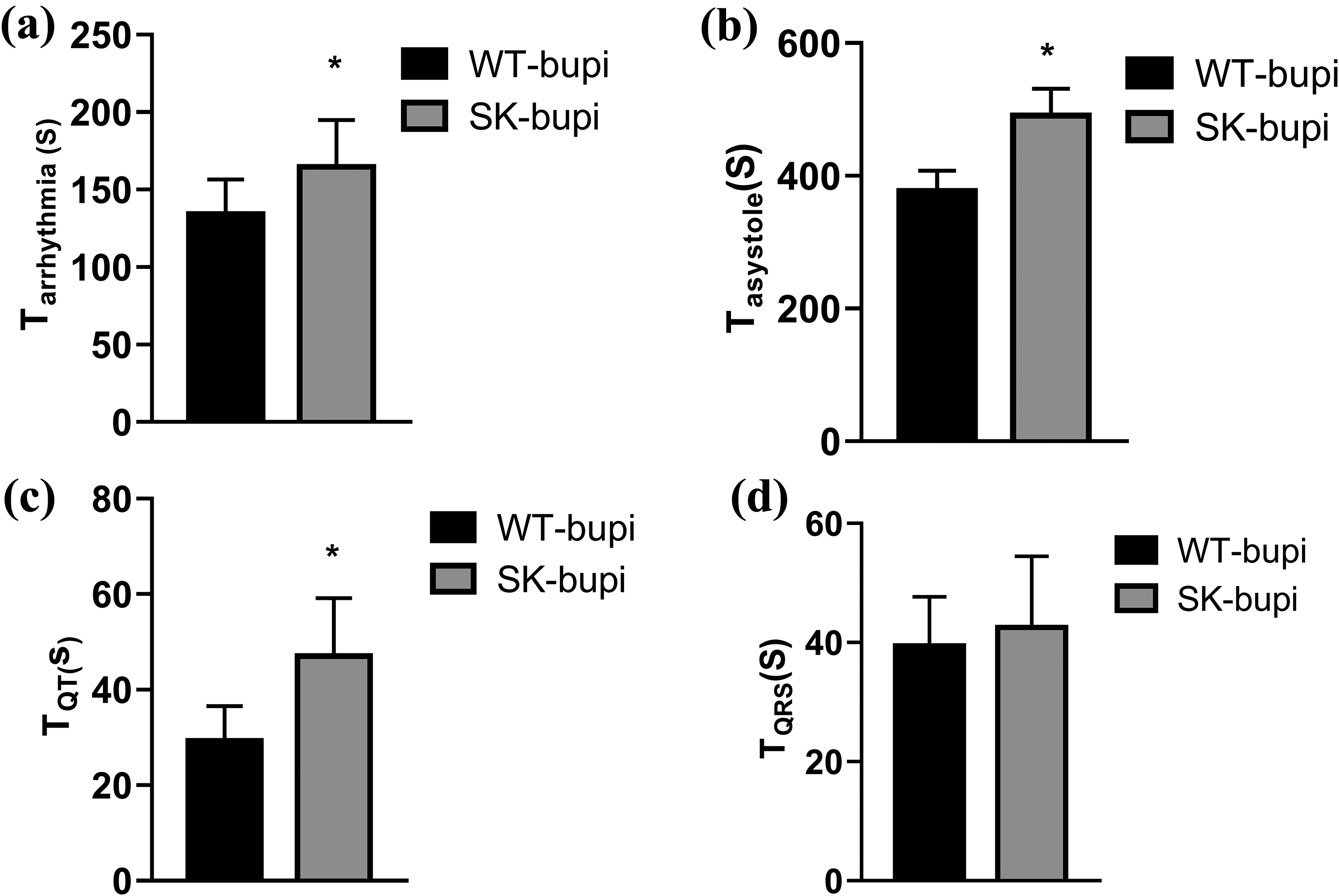
Time to the occurrence of ECG events after bupivacaine infusion. (a) Tarrhythmia values between WT-bupi and SK-bupi groups; (b) Tasystole values between WT-bupi and SK-bupi groups; (c) TQT values between WT-bupi and SK-bupi groups; (d) TQRS values between WT-bupi and SK-bupi groups. WT-bupi: infusion of bupivacaine in WT mice; SK-bupi: infusion of bupivacaine in SK2 gene knockout mice; Tarrhythmia: time to arrhythmia after infusion of bupivacaine; Tasystole: time to cardiac asystole after infusion of bupivacaine; TQT: time to QT prolongation by 20% after bupivacaine infusion; TQRS: the time to QRS wave widening by 20% after bupivacaine infusion; * P < 0.05 between WT-bupi and SK-bupi groups.
TQT and TQRS
The QT interval and QRS wave width were gradually increased after the infusion of bupivacaine in both the SK-bupi group and the WT-bupi group. The TQT in the SK-bupi group was longer than in the WT-bupi group (P < 0.05), while TQRS was comparable in both groups (Figure 2(c) and (d)). The QT interval and QRS wave width were not prolonged in the SK and WT groups.
HR and MAP values after bupivacaine infusion
After bupivacaine infusion, cardiac asystole occurred in all mice, albeit at different time points. At 2–6 minutes after the infusion of bupivacaine (T2–T6), HR in the SK-bupi group was significantly higher than in the WT-bupi (P < 0.05) but was similar in SK and WT groups (P > 0.05) (Figure 3). The time to 25%, 50%, and 75% reduction in HR was significantly longer in the SK-bupi group than in the WT group (all P < 0.05) (Table 1).

Heart rate after bupivacaine infusion. WT: infusion of normal saline in WT mice; SK: infusion of bupivacaine in SK2 knockout mice; WT-bupi: infusion of bupivacaine in WT mice; SK-bupi: infusion of bupivacaine in SK2 gene knockout mice; HR: heart rate; *P < 0.05, between WT-bupi and SK-bupi groups.
The MAP values were comparable in the SK-bupi and WT-bupi groups (Figure 4). Moreover, there was no significant difference in the time to 25%, 50%, and 75% reduction in MAP between the SK-bupi group and WT-bupi group (all P > 0.05) (Table 2).

Mean arterial pressure after bupivacaine infusion. WT: infusion of normal saline into WT mice; SK: infusion of bupivacaine in SK2 knockout mice; WT-bupi: infusion of bupivacaine in WT mice; SK-bupi: infusion of bupivacaine in SK2 gene knockout mice; MAP: mean arterial pressure. *P < 0.05, between WT-bupi and SK-bupi groups.
Time-dependent changes in cardiac function parameters after bupivacaine infusion.
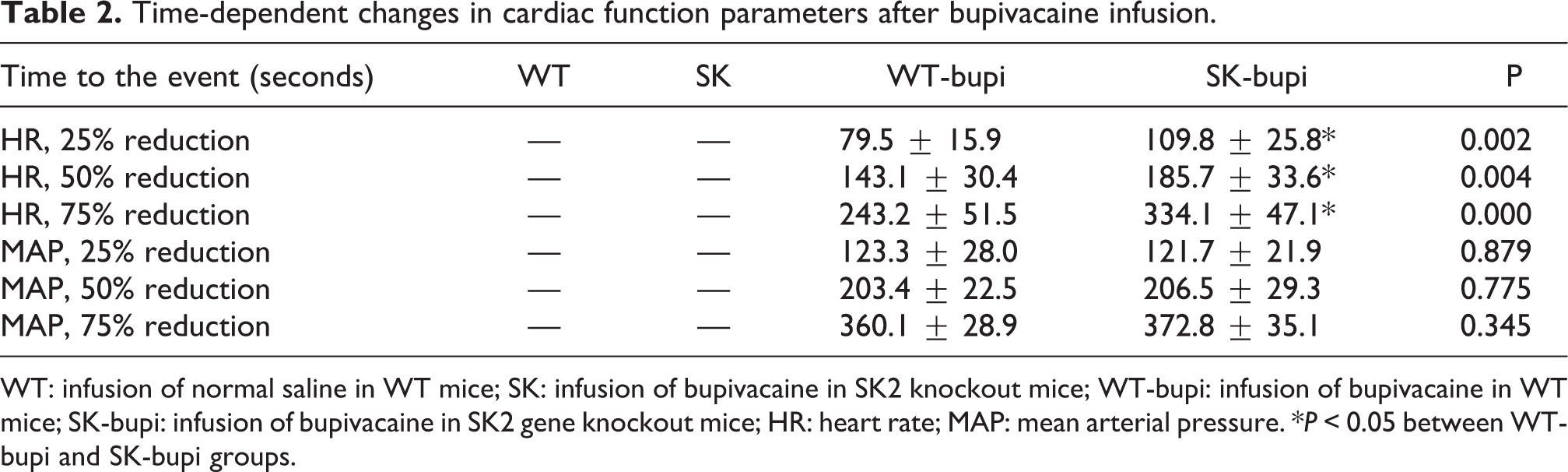
WT: infusion of normal saline in WT mice; SK: infusion of bupivacaine in SK2 knockout mice; WT-bupi: infusion of bupivacaine in WT mice; SK-bupi: infusion of bupivacaine in SK2 gene knockout mice; HR: heart rate; MAP: mean arterial pressure. *P < 0.05 between WT-bupi and SK-bupi groups.
Bupivacaine in plasma and myocardium
The concentration of bupivacaine in the plasma and the myocardium was significantly higher in the SK-bupi group than in the WT-bupi group (both P < 0.05) (Figure 5).

Bupivacaine concentrations in plasma and myocardium of mice. (a) Cplasmavalues between WT-bupi and SK-bupi groups; (b) Ctissue values between WT-bupi and SK-bupi groups. WT-bupi: infusion of bupivacaine in WT mice; SK-bupi: infusion of bupivacaine in SK2 gene knockout mice; CPlasma: the concentration of bupivacaine in plasma; CTissue: the bupivacaine content in the heart. *P < 0.05, between WT-bupi and SK-bupi groups.
Discussion
The main finding of the present investigation is that SK2 gene knockout delays the time of the onset of manifestations of cardiac toxicity such as arrhythmia and cardiac arrest in mice. Additionally, the tolerance to bupivacaine in SK2 gene knockout mice was increased. In this study, arrhythmia was defined as the occurrence of the first dropped beat. 17
It is generally accepted that bupivacaine cardiotoxicity results from a widespread block of cardiac ion channels. 6 –8 SK2 channel, as a channel widely distributed in the heart, plays an important role in cardiac electrophysiology.10,11 Previous study has shown that bupivacaine could inhibit the SK2 current in a concentration-dependent manner in HEK293 cells. 15 After knocking out the SK2 channel gene, the toxicity of bupivacaine in an isolated mouse heart was significantly delayed. To verify the results, an in vivo mouse model was used in this study. The present study demonstrated that the time to arrhythmia and cardiac asystole in SK2 knockout mice was significantly longer than in wild-type mice. This finding suggests that the deletion of the SK2 channel could also delay the occurrence of bupivacaine-induced cardiotoxicity in vivo. Our study provided further evidence that the SK2 channel mediates bupivacaine cardiotoxicity.
It has been shown that bupivacaine could prolong QT interval and increase the width of the QRS wave.18,19 Our results are consistent with previous studies.18,19 Interestingly, the present work also found that the time to the prolongation of the QT interval by 20% was longer in SK2 knockout mice than in WT mice. This result can be explained by the fact that a low concentration of bupivacaine can significantly inhibit SK2 current in WT mice, leading to the prolongation of the QT interval. This may be one of the reasons for the higher susceptibility to cause arrhythmia and cardiac asystole in WT mice. The block of the SK2 channel by bupivacaine produces ECG changes, which may be involved in the process of bupivacaine cardiotoxicity.
The present study has also shown that the concentration of bupivacaine in plasma and myocardium of SK2-knockout mice at the time of cardiac asystole was significantly higher than in the WT mice. This result indicated that the knockout of the SK2 gene could enhance the tolerance of the mouse heart to bupivacaine. As an important ion transporter, the SK2 channel in myocytes is involved in electrophysiological conduction rather than myocardial contractility. 11 This study also found that the heart rate in SK2 knockout mice decreased slowly when bupivacaine was infused, but the mean arterial pressure. This result further confirmed that the bupivacaine-induced block of the SK2 current is involved in bupivacaine cardiotoxicity, although it may not be the primary reason.
Frequently, the organism does not exhibit an obvious phenotype after gene knockout because compensatory pathways can be often activated.20,21 The results showed no evidence of arrhythmia or changes in the QT and QRS intervals in the knockout mice. The absence of differences in arrhythmia and hemodynamics between the SK and WT groups throughout the course of the experiment indicates that the mice activated a certain functional compensation after gene knockout. However, the compensatory mechanism remains unknown.
Limitations
Mice weighing approximately 25 g were selected for this study. The blood volume of mice could change easily in the presence of bleeding during the operation. To eliminate this factor, the cardiac venous pressure was monitored and controlled at 5–10 mmHg before the infusion of bupivacaine. Lactate Ringer’s solution was infused slowly when the central venous pressure decreased to 5 mmHg. After the infusion of bupivacaine, the effect of fluid volume on hemodynamics could be ignored because the velocity of the entering fluid was the same in all groups.
Conclusion
The knockout of the SK2 channel can reduce the sensitivity to bupivacaine-induced cardiotoxicity in the mouse. This study advances our understanding of the mechanism of bupivacaine-induced cardiotoxicity.
Footnotes
Acknowledgment
We would like to thank Dr Yun Xia of the Ohio State University Medical Center for the critical reading of the manuscript.
Declaration of conflicting interests
The author(s) declare no potential conflicts of interest with respect to the research, authorship, and publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by the National Natural Science Foundation of China, Beijing, China (Grant No: 82003876) and the National Natural Science Foundation of Zhejiang Province, China (Grant No: LQ18H090006).