
Abstract
Methylphenidate (MPH) is used as the first-line treatment for attention-deficit hyperactivity disorder. However, there are concerns that this treatment may be associated with increased risk of retinal damage. This study was to investigate cytotoxicity of MPH on photoreceptor cells and explore its underlying mechanisms. MPH-caused cell toxicity was established in 661 W cells. Cytotoxicity was evaluated by 3-(4,5-dimethylthiazol)-2,5-diphenyltetrazolium-bromid and lactate dehydrogenase assays. Oxidative stress was measured by the markers: glutathione (GSH) reductase, catalase, and superoxide dismutase activities as well as GSH, reactive oxygen species, and malondialdehyde levels. Gene and protein expression was detected by real-time polymerase chain reaction (PCR) and western blot, respectively. Results showed that MPH decreased 661 W cell viability, increased caspase-3/9 activities, and induced oxidative stress. Furthermore, MPH treatment increased messenger RNA (mRNA) expression of Beclin-1 and microtubule-associated protein 1A/1B-light chain 3B (LC3B) protein expression in 661 W cells, suggesting autophagy was induced. MPH treatment also upregulated p-JAK1/p-STAT1 protein expression. These data demonstrated that MPH could increase oxidative stress in photoreceptor cells to cause cell toxicity via autophagy, providing the scientific rationale for the photoreceptor cell damage caused by the MPH administration.
Introduction
As a piperidine-derived central nervous system stimulant, methylphenidate (MPH, Ritalin) is widely used to treat attention-deficit hyperactivity disorder (ADHD) in children. 1 However, there are concerns that MPH is being diverted from legitimate use among teens and college students. The retinopathy with Ritalin is more common among abusers because abusers crush the tablets, prepare an aqueous suspension, and inject it. So in most of the cases, the retinopathy is induced by insoluble fillers, talc, and cornstarch that are used in tablets. 2 Toxicity of MPH is similar to toxicity with other amphetamine-like drugs. Neurologic effects include agitation, euphoria, dizziness, hallucinations, delusions, psychosis, lethargy, seizures, tremors, and hyperreflexia. 3,4 Large dose of MPH may cause cataract and glaucoma. 5 Other ocular side effects include dry eyes, mydriasis, decreased visual acuity, and blurry vision. 6 However, the toxic effects of MPH on the retina have not been completely elucidated.
The mechanism of action of MPH is related to its action on multiple neurotransmitters including dopamine (DA) and serotonin. The activity on dopaminergic areas of the brain is believed to be most responsible for its effects on hyperactivity and behavior. Acute increases in cytoplasmic and extracellular DA contribute to oxidatively damaged axon terminals. 7 Studies showed that methamphetamine-induced cytotoxicity was associated with excessive production of oxygen-based free radicals, causing neuronal death through apoptosis. 8,9 Treating rats with methamphetamine repeatedly resulted in the imbalance of antioxidative/oxidative system in retinas, and the resultant increased oxidative stress may play a pivotal role in the pathophysiology of retinal degenerative events and impair the normal function of the visual system. 10 MPH has been known to have psychostimulus effects similar to methamphetamine. Therefore, we hypothesize that MPH also caused toxicity through oxidative stress. Moreover, both methamphetamine and MPH induced astrocytic activation in limbic neuron/glia cocultures. 11 It is well known that activated astrocytes and microglia produce not only reactive oxygen species (ROS) that are harmful to neurons but also pro-inflammatory cytokines to propagate the neuroinflammatory cascade potentially amplifying the neuron damage. 12 Neuroinflammation has been implicated as an important mechanism associating with a number of neuropsychological impairments. 13
Although a study showed that application of MPH alone exerted no neurotoxic, but neuroprotective effects in vitro. 14 Effects of MPH on photoreceptor cells have not been examined. This study was to investigate effects of MPH on 661 W cells and explore the underlying mechanisms.
Materials and methods
Cell line, chemicals, and reagents
Cell line (661 W) was purchased from Beijing Cell bank (Beijing, China). RPMI1640 (Roswell Park Memorial Institute) medium and fetal bovine serum (FBS) were purchased from Gibco (Gaithersburg, Maryland, USA). 3-(4,5-dimethylthiazol)-2,5-diphenyltetrazolium-bromid (MTT) and Tofacitinib were purchased from Sigma-Aldrich (St. Louis, Missouri, USA). Lactate dehydrogenase (LDH), glutathione reductase (GR), catalase, superoxide dismutase (SOD), glutathione (GSH), and malondialdehyde (MDA) assay kits were purchased from Jiancheng Biological Engineering (Nanjing, China). 2′,7′-Dichlorodihydrofluorescein diacetate (H2DCFDA) were purchased from Molecular Probes (Eugene, Oregon, USA). Caspase-3 and caspase-9 activity assay kits were purchased from Kaiji Institute of Biological Engineering (Nanjing, China). 3,3′-Dihexyloxacarbocyanine iodide (DiOC6(3)) was purchased from ThermoFisher Scientific (Waltham, Massachusetts, USA). Caspase-3 inhibitor Z-DEVD-FMK and caspase-9 inhibitor Z-LEHD-FMK were purchased from Merck (Billerica, Massachusetts, USA). Real-time PCR reagents were purchased from ThermoFisher. All solvents and chemicals were of analytical grade and purchased from Sinopharm Co. Ltd. (Shanghai, China). MPH (CAS 113-45 -1) was purchased from National Institutes for Food and Drug Control (Beijing, China) and dissolved in phosphate-buffered saline.
Cell culture and treatment
Cells (661 W) were placed into 75 T tissue flasks and cultured at 37°C under the humidified 5% CO2 atmosphere in RPMI1640 medium supplemented with 10% FBS, 100 U/ml penicillin, and 100 μg/ml streptomycin. Cells (661 W) were treated with MPH at different concentrations (10, 30, and 90 µM) for 48 h. The concentration of MPH was based on clinical study. With extrapolation using the maximum clinical oral quantity of 60 mg MPH as the administered dosage, one could expect to see as much as 30 mg in the brain (approximately 103 mM). 15 Cells treated with vehicle were used as the negative control. To investigate the signaling pathway, 661 W cells were individually pretreated with 20 μM Z-DEVD-FMK, 20 μM Z-LEHD-FMK, or 10 μM Tofacitinib for 2 h prior to exposure to MPH. Cells treated with vehicle or inhibitor alone were used as control.
Cell viability measurement
Cell viability was determined by MTT assay. After treatment, 661 W cells were plated in 96-well plates. Ten-microliter MTT solution at 0.5 mg/ml was added and incubated for 4 h at 37°C. The formazan crystals were solubilized with 100 µl dimethyl sulfoxide and the absorbance was measured at 570 nm by the microplate reader. Cell viability was expressed as the percentage of the negative control, which was set to 100%.
LDH leakage assay
LDH leakage was measured by the LDH assay kit. After treatment, the culture medium was collected and centrifuged at 3000 r/min at 4°C for 10 min. The activity of LDH in the supernatant was measured according to the manufacturer’s instructions. The LDH leakage was expressed as the percentage of the negative control, which was set to 100%.
MMP measurement
After treatment, 661 W cells were harvested and resuspended in 500 µl of 100 nM DiOC6(3) at 37°C for 30 min. Mitochondrial membrane potential (MMP) was measured by flow cytometry for mean fluorescence intensity (MFI). The MFI was expressed as the percentage of the negative control, which was set to 100%.
Measurement of caspase activity
After treatment, 661 W cells were washed twice and lyzed. Caspase-3/9 activities were measured by the assay kits according to the manufacturer’s instructions. Activities of caspase-3/9 were expressed as the percentage of the negative control, which was set to 100%.
ROS measurement
H2DCFDA was used to measure ROS production in 661 W cells. After treatment, cells were incubated with H2DCFDA at a final concentration of 10 µM for 20 min at 37°C and then washed twice. ROS were measured using a fluorescence microplate reader with excitation and emission wavelengths at 488/525 nm.
GR, catalase, SOD activities, GSH, and MDA levels
After treatment, 661 W cells were homogenized in lysis buffer (ReadyPrep Protein Extraction Kit, 1632086, Bio-Rad, Hercules, California, USA) according to the manufacturer’s instructions. The supernatant of cell lysate was adjusted to be 1 mg protein/ml to measure GR, catalase, and SOD activities as well as GSH and MDA levels according to manufacturers’ instructions.
Real-time PCR
After treatment, total RNA was extracted from 661 W cells. Complementary DNA was generated using 0.5 µg total RNA with SuperScript master mix kit (Bio-Rad). Quantitative PCR was performed using SYBR Green Supermix (ThermoFisher). The expression of genes of interest in different samples was quantified by comparative C t value method. The mRNA levels were normalized to the housekeeping gene β-actin. The gene-specific primer sequences were the following. For Beclin-1, forward: cagtgttcctgtggagtgga and reverse: tgcacacagtccagaaaagc, NCBI reference: NM_019584.4 and for β-actin, forward: agccatgtacgtagccatcc and reverse: gctgtggtggtgaagctgta, NCBI reference: NM_007393.5.
Western blot analysis
After treatment, 661 W cells were harvested and the protein was collected. The protein concentration was measured using a Bio-Rad Protein Assay Kit. Protein (40 μg) from each sample was subjected to electrophoresis on a 12% (v/v) sodium dodecyl sulfate –polyacrylamide gel and then transferred to the Immobilon-P polyvinylidene difluoride membrane. The membrane was blocked with 2% bovine serum albumin at room temperature for 1 h and incubated with indicated primary antibodies (anti-cleaved caspase-3 rabbit pAb ab49822, 1:500; anti-caspase-3 rabbit pAb ab13847, 1:500; anti-cleaved caspase-9 rabbit mAb #9509, 1:1000; anti-caspase-9 rabbit mAb ab202068, 1:2000; anti-nicotinamide adenine dinucleotide phosphate (NADPH) oxidase 4 rabbit pAb MBS176126, 1:1000; anti-heme oxygenase (HO)-1 rabbit mAb ab189491, 1:2000; anti-LC3B rabbit pAb ab51520, 1:1000; anti-JAK1 (phospho Tyr1034/1035) rabbit pAb #3331, 1:1000; anti-JAK1 rabbit pAb ab47435, 1:1000; anti-STAT1 (phospho Y701) rabbit pAb ab30645, 1:1000; anti-STAT1 rabbit pAb ab47425, 1:1000; and anti-β-actin, rabbit pAb, ab15263, 1:3000) at 4°C overnight. Thereafter, blots were probed with horseradish peroxidase (HRP)-conjugated secondary antibody (goat anti-rabbit IgG H&L HRP, ab205718, 1:3000) for 1 h. After incubation, membrane was washed three times, and the antigen–antibody complexes were visualized by the enhanced chemiluminescence system (PerkinElmer, Waltham, Massachusetts, USA).
Statistical analysis
Data were expressed as mean ± standard deviation. Kolmogorov–Smirnov test was used for the normality test. Data were analyzed by one-way analysis of variance followed by Dunnett’s test using SPSS 16.0 software (SPSS, Chicago, Illinois, USA). A p-value <0.05 was considered statistically significant.
Results
MPH caused 661 W cell toxicity
Cells (661 W) were treated with MPH at different concentrations. Comparing to the negative control, obvious toxicity on 661 W cells caused by MPH was observed, shown by MTT assay (regression coefficient: −0.9985, p < 0.05), together with the increasing leakage of LDH (regression coefficient: 0.9904, p < 0.05) in a concentration-dependent manner. Moreover, MPH significantly decreased the MMP in 661 W cells (regression coefficient: −0.9916, p < 0.05) in a concentration-dependent manner (Figure 1).
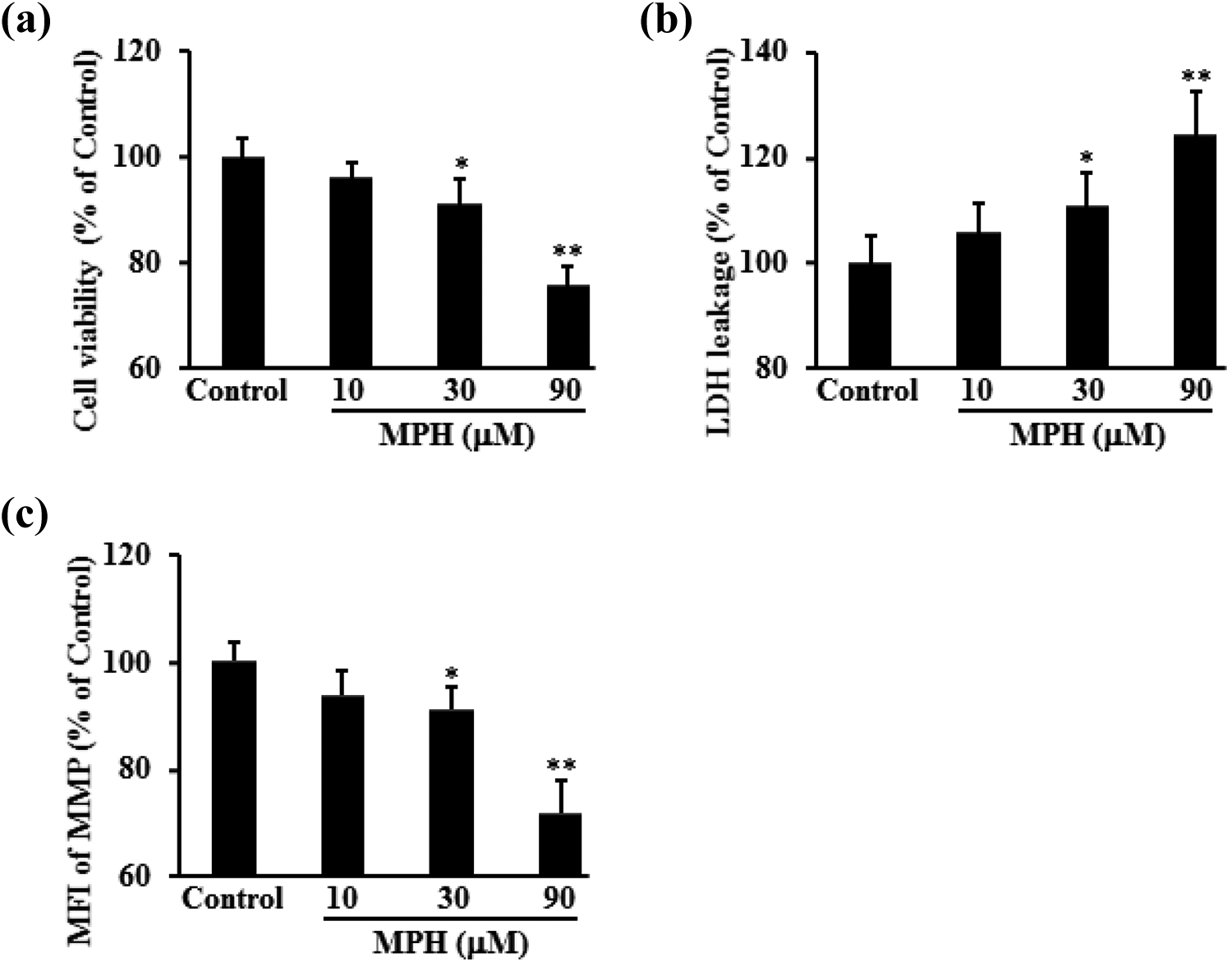
MPH caused toxicity in 661 W cells. Cells (661 W) were treated with MPH at 10, 30, and 90 µM for 48 h. The cell viability was tested by (a) MTT assay, (b) LDH leakage assay, and (c) mitochondrial membrane potential. Data were expressed as mean ± SD. *p < 0.05; **p < 0.01 versus control. Experiments were performed three times independently. MPH: methylphenidate; MTT: 3-(4,5-dimethylthiazol)-2,5-diphenyltetrazolium-bromid; LDH: lactate dehydrogenase; SD: standard deviation.
MPH increased activities and expression of caspase-3/9 in 661 W cells
MPH exposure significantly increased caspase-3/9 activities in a concentration-dependent manner (regression coefficient: 0.9860, p < 0.05 for caspase-3 and regression coefficient: 0.9888, p < 0.05 for caspase-9). To confirm whether caspase-3/9-dependent signaling was the main contributor to MPH-induced cytotoxicity, specific inhibitors were applied prior to MPH exposure. Our results demonstrated that caspase inhibitors Z-LEHD-FMK or Z-DEVD-FMK significantly abolished MPH-induced cytotoxicity (p < 0.01). Moreover, MPH increased the cleaved caspase-3/9 protein expression (regression coefficient: 0.9897, p < 0.05 for cleaved caspase-3 and regression coefficient: 0.9853, p < 0.05 for cleaved caspase-9), while not changing the total caspase-3/9 protein expression (regression coefficient: −0.2009 for caspase-3 and regression coefficient: 0.0626 for caspase-9; Figure 2).
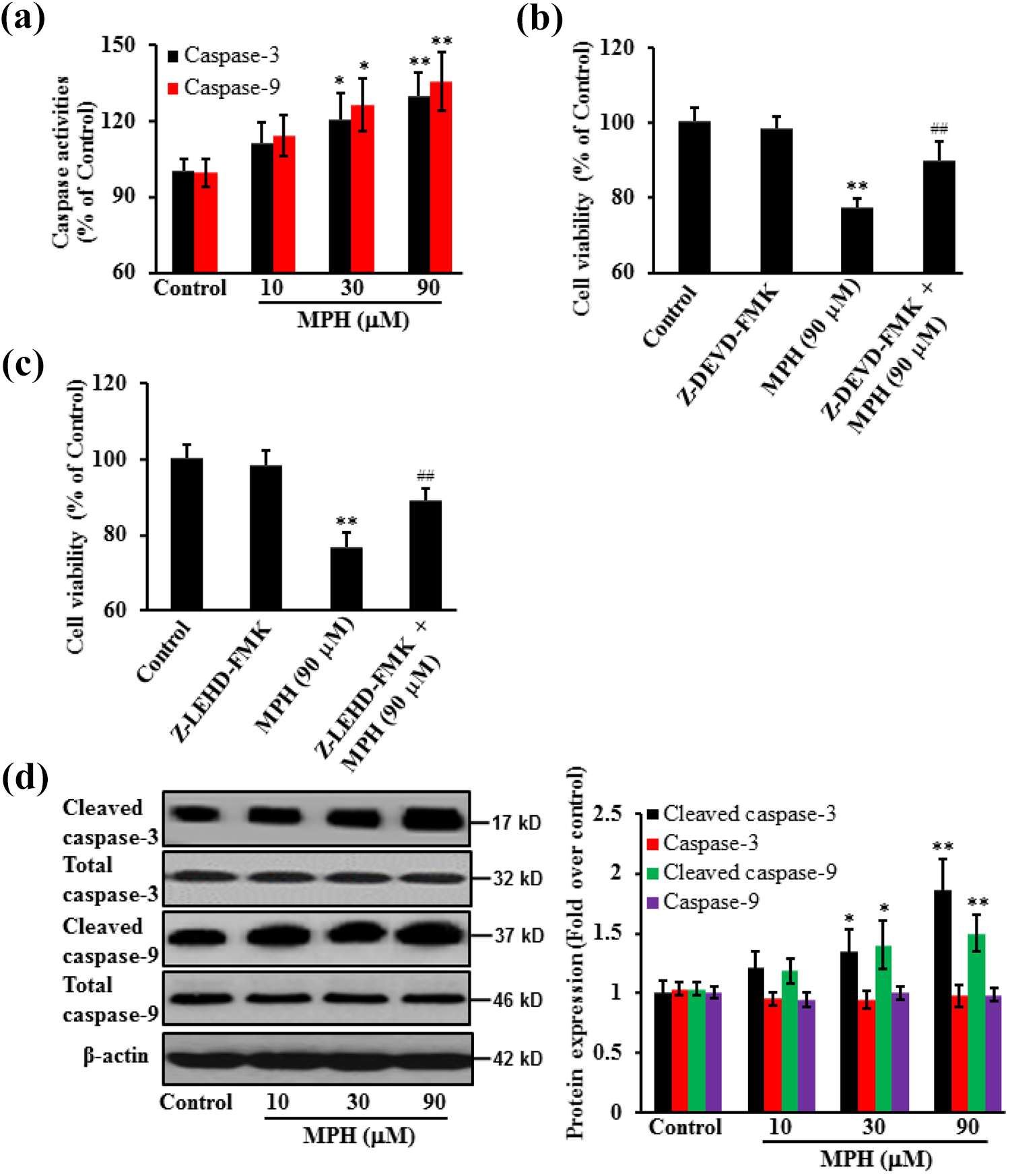
Effects of MPH on caspase-3/9 in 661 W cells. Cells (661 W) were treated with MPH at 10, 30, and 90 µM for 48 h. (a) MPH treatment significantly increased caspase-3/9 activities in cell lysates. (b) and (c) Pretreatment with Z-DEVD-FMK (caspase-3 inhibitor, 20 μM) or Z-LEHD-FMK (caspase-9 inhibitor, 20 μM) for 2 h largely abolished MPH-reduced cell viability. (d) Moreover, MPH increased the protein expression of cleaved caspase-3/9, while not changing the protein expression of total caspase-3/9. *p < 0.05; **p < 0.01 versus control; ## p < 0.01 versus MPH alone. Experiments were performed three times independently. MPH: methylphenidate.
MPH increased the oxidative stress in 661 W cells
Levels of ROS, MDA, and GSH as well as activities of GR, catalase, and SOD were measured after MPH treatment. Comparing to the control, oxidative stress significantly increased in 661 W cells after MPH treatment, shown by increased ROS level (regression coefficient: 0.9818, p < 0.05) and MDA content (regression coefficient: 0.9721, p < 0.05) and decreased GSH content (regression coefficient: −0.9843, p < 0.05) and activities of SOD (regression coefficient: −0.9769, p < 0.05), catalase (regression coefficient: −0.9805, p < 0.05) as well as GR (regression coefficient: −0.9725, p < 0.05) in a concentration-dependent manner. To further confirm the mechanism, NADPH oxidase 4, a major enzyme responsible for production of superoxide by transferring electrons across the membrane from NAD(P)H to molecular oxygen, 16 and HO-1, a cell-protective enzyme, were detected by western blot. Results shown that MPH significantly increased the protein expression of NADPH oxidase 4 (regression coefficient: 0.9931, p < 0.05) and decreased the protein expression of HO-1 (regression coefficient: −0.967, p < 0.01) in a concentration-dependent manner, indicating MPH caused cell damage through increasing the oxidative stress (Figure 3).
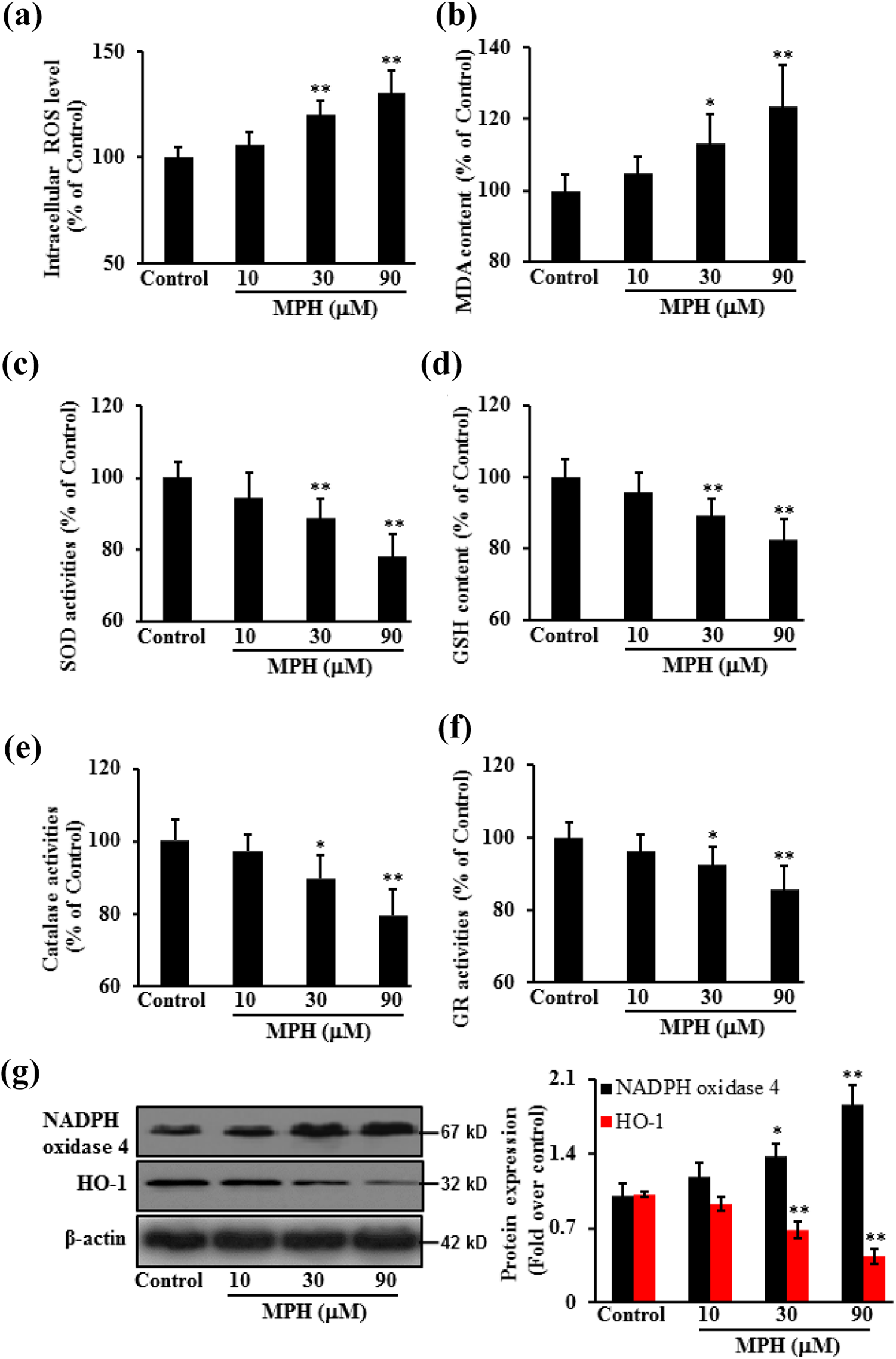
MPH increased oxidative stress in 661 W cells. Cells (661 W) were treated with MPH at 10, 30, and 90 µM for 48 h. Cells were collected to measure the oxidative stress markers: (a) intracellular ROS level; (b) MDA content; (c) SOD activity; (d) GSH level; (e) catalase activity; (f) GR activity; and (g) NADPH oxidase 4 and HO-1 protein expression. Data were expressed as mean ± SD. *p < 0.05; **p < 0.01 versus control. Experiments were performed three times independently. MPH: methylphenidate; ROS: reactive oxygen species; MDA: malondialdehyde; SOD: superoxide dismutase; GSH: glutathione; GR: glutathione reductase; NADPH: nicotinamide adenine dinucleotide phosphate; HO: heme oxygenase; SD: standard deviation.
MPH increased autophagy in 661 W cells
Effects of MPH on 661 W cell autophagy were explored by examining the mRNA expression of Beclin-1 and protein level of LC3B. Results showed that MPH increased Beclin-1 mRNA (regression coefficient: 0.9985, p < 0.05) and LC3B protein expression (regression coefficient: 0.9785, p < 0.05) in 661 W cells in a concentration-dependent manner (Figure 4).

MPH induced autophagy in 661 W cells. Cells (661 W) were treated with MPH at 10, 30, and 90 µM for 48 h. Cells were collected to measure the mRNA and protein expression: (a) Beclin-1 mRNA expression and (b) LC3B protein expression. Data were expressed as mean ± SD. *p < 0.05; **p < 0.01 versus control. Experiments were performed three times independently. MPH: methylphenidate; mRNA; messenger RNA; SD: standard deviation.
MPH increased the protein expression of p-JAK1 and p-STAT1 in 661 W cells
To identify the action mechanisms of MPH, the protein expression of JAK1/STAT1 was examined. The relative protein expression of p-JAK1/p-STAT1 markedly increased (regression coefficient: 0.9895, p < 0.05 for p-JAK1 and regression coefficient: 0.9853, p < 0.05 for p-STAT1) after MPH treatment in a concentration-dependent manner, while not changing the total JAK1/STAT1 (regression coefficient: −0.2343 for JAK1 and regression coefficient: −0.1862 for STAT1) compared with the control. However, the increased expression levels of p-JAK1 and p-STAT1 were repressed by the specific inhibitor Tofacitinib (p < 0.05; Figure 5).

MPH increased p-JAK1/p-STAT1 protein expression. Cells (661 W) were treated with MPH at 10, 30, and 90 µM for 48 h. Cells were collected to measure the protein expression. (a) MPH treatment increased the protein expression of p-JAK1/p-STAT1, while not changing the protein expression of JAK1/STAT1 and (b) Tofacitinib (JAK/STAT pathway inhibitor) repressed the upregulation of p-JAK1/p-STAT1 caused by MPH. *p < 0.05; **p < 0.01 versus control; # p < 0.05; ## p < 0.01 versus Tofacitinib alone. Experiments were performed three times independently. MPH: methylphenidate.
Discussion
MPH is a popular drug in the treatment of ADHD in clinical practice. Although changes in visual system have been reported as adverse effects, including cataract, glaucoma, and visual loss, 5,6 there is little scientific evidence about the effects of this drug on the retina. To quantify toxicity of MPH on the retina, this study examined the effects of various concentrations of MPH on photoreceptor cells using the 661 W cell line.
As one of sympathomimetic amine stimulants, MPH is structurally related to amphetamine with effects on dopaminergic and noradrenergic systems. 17 MPH blocks the DA transporters and application of 1–1000 µM MPH to striatum in awake rats increased extracellular DA in a dose-dependent manner, 15,18 implying that its action is associated with increases in DA concentrations. MPH was also reported to exert the degenerative effects on cornea through activating dopaminergic mechanism via similar neuronal paths to induce morphological changes. 19 DA regulates retinal morphogenesis by inhibiting the extension of neuritis of retinal cells. Moreover, retinal dopaminergic neurons are involved in controlling the organization of ganglion cells and modulation of the physiological activities of photoreceptors. 20,21 Our results demonstrated that MPH caused toxicity in 661 W cells. Effects of MPH were further investigated by exploring the intrinsic apoptotic mechanisms. The decrease of MMP has been considered to regulate apoptotic cell fate because mitochondrial function is one of the apoptotic mediators that elicit intrinsic signaling. 22,23 Our data showed that MPH significantly decreased MMP and increased caspase-3/9 activities as well as protein expression.
It is commonly accepted that the principal source of ROS in the cell is the mitochondrial respiratory chain. 24 ROS is one of the major sources of DNA damage as it could directly modify the DNA or indirectly generate different lesions, both affecting cell viability. 25 The antioxidant enzymes SOD, catalase, and GR are responsible for the removal of ROS in general. They are present in all cellular districts and act in concert with other antioxidants (e.g. GSH) to fully scavenge ROS. 26,27 MPH was reported to cause anxiety-like behavior in animals after administration, 28 associated with oxidative stress. 29,30 In addition, it has been shown that MPH abuse causes neurotoxicity by reducing the activity of antioxidative enzymes like SOD and converting the protective form of GSH into the damaging form of glutathione disulfide (GSSG). 31,32 These findings are in accordance with our study, which has confirmed MPH-induced oxidative stress in 661 W cells.
A growing amount of evidence showed ROS as early inducers of autophagy upon nutrient deprivation. 33 Treatment with antioxidants prevents autophagy, suggesting that redox imbalance has an important role in driving the process. The evidence that the sole chemically induced oxidation of GSH is able to induce autophagy even in the absence of any autophagic stimulus underlines the importance of redox homeostasis in autophagy commitment. 34 Our results showed that exposing 661 W cells to MPH caused autophagy, evidenced by a significant increase in the expression of Beclin-1 gene and LC3B protein. Studies have demonstrated that oxidative stress utilized the JAK/STAT signal pathway to induce apoptosis or cell proliferation. 35 JAK/STAT signaling pathway participates in a variety of pathological processes and regulating JAK/STAT can alleviate cell damage caused by oxidative stress. 36 Activation of JAK1/STAT1 signaling was required for increased expression of Beclin-1, a key regulator of autophagy, 37 which is consistent with our results showing that MPH treatment significantly increased the protein expression of p-JAK1/p-STAT1 in 661 W cells.
Some study limitations should be mentioned. Firstly, this study used cell line as the in vitro model, which is far from the real situation and can only be used as the lead compound screening. Its activities need to be confirmed by animal models. Secondly, the exact mechanisms of MPH on photoreceptor cell damage should be further explored. Finally, it is uncertain whether these data accurately reflect molecular changes in human.
In conclusion, our study demonstrated that although MPH seems to be a safe option for the ADHD treatment, it should be used with caution, since treatment of 661 W cells with MPH resulted in cell damage. This suggests possible cytotoxic effects of MPH on photoreceptor cells, providing the scientific rationale to minimize risks when this drug is taken.
Footnotes
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This study was supported by grants from Inner Mongolia Natural Science Foundation (2019MS08142, 2013MS11109, 2011MS1137).