
Abstract
As an extremely addictive psychostimulant drug and an illicit dopaminergic neurotoxin, methamphetamine (METH) conducts to enhance satisfaction, feelings of alertness through influencing monoamine neurotransmitter systems. Long-lasting exposure to METH causes psychosis and increases the risk of neurodegeneration. 6-Formyl-5-isopropyl-3-hydroxymethyl-7-methyl-1H-indene (FIHMI) is a novel compound with potent antioxidant properties. This study was to investigate whether FIHMI could mitigate METH-induced photoreceptor cell toxicity. METH-caused cell toxicity was established in 661W cells and protective effects of FIHMI at different concentrations (1–10 µM) was examined. FIHMI significantly attenuated the METH-caused cell damage in 661W cells, evidenced by increasing cell viability and mitochondrial membrane potential, decreasing cytochrome c release and DNA fragmentation, inhibiting activities of caspase 3/9, and changing expression of apoptosis-related protein. Furthermore, FIHMI treatment decreased mRNA expression of Beclin-1 and LC3B protein expression in METH-induced 661W cells suggesting autophagy is reduced. FIHMI decreased the oxidative stress through increasing protein expression of nuclear factor (erythroid-derived 2)-like 2. These data demonstrated FIHMI could inhibit oxidative stress, which may also play an essential role in the regulation of METH-triggered apoptotic response, providing the scientific rational to develop FIHMI as the therapeutic agent to alleviate METH-induced photoreceptor cell toxicity.
Keywords
Introduction
Methamphetamine (METH) is an illicit dopaminergic neurotoxin and potent psychostimulant drug with wide ranging effects on the central nervous system through influencing on monoamine neurotransmitter systems. 1,2 METH users dramatically increased more than 150% over the past 5 years. 3 METH enhances the motor activity, happiness, satisfaction, and consciousness but decreases appetite. Studies have shown that long-term METH abuse causes neurodegeneration by neuroimaging analyses, including damage to dopamine axons, gray matter atrophy followed by hypertrophy of the white matter. 4,5 Chronic exposure or taken in binge doses, METH can cause irreversible damage to photoreceptor cells. 6 Studies showed METH damaged monoamine-containing nerve terminals and generated an imbalance in the release and reuptake of dopamine leading to neurological abnormalities. 7,8 However, the underlying molecular mechanisms of METHs toxicity in degeneration remain to be interpreted.
Reports have shown that METH application generates reactive oxygen species (ROS). 9 Repeated treatment with METH to rats caused imbalance of oxidative/anti-oxidative system in retina and the compromised antioxidant defense may interfere with the normal functioning of the visual system and play a role in the pathophysiology of ocular degenerative events that are linked to increased oxidative stress. 10 On the other hand, METH can also promote the production of inflammatory factors, resultantly increasing apoptosis. 11 Understanding the mechanisms involved in the toxicity of METH can lead to the discovery of new strategies to prevent degenerative process.
Different antioxidants have been used to alleviate the oxidative damage to treat disorders. 12 6-Formyl-5-isopropyl-3-hydroxymethyl-7-methyl-1H-indene (FIHMI) is a new compound isolated from the leaves of Micromelum integerrimum 13 with potent antioxidant activities. This study was to explore the protective effects and potential mechanisms of FIHMI in METH-induced toxicity of 661W cells.
Materials and methods
Reagents
661W cell line was from Beijing Cell Bank (Beijing, China). RPMI1640 culture medium was from Gibco BRL (Waltham, Massachusetts, USA). 3-(4,5-Dimethylthiazol)-2,5-diphenyltetrazolium bromide (MTT) was purchased from Sigma–Aldrich (St Louis, Missouri, USA). Cytochrome c assay kit was from R&D systems (Minneapolis, Minnesota, USA). 5,5′,6,6′-Tetrachloro-1,1′,3,3′-tetraethylbenzimidazolylcarbocyanine iodide (JC-1) and 2′,7′-dichlorofluorescin diacetate (DCF-DA) were purchased from Molecular Probes (Eugene, Oregon, USA). Caspase 3/9 activity assay kits were from Promega (Madison, Wisconsin, USA). Cell Death Detection ELISAplus kit was from Roche Applied Sciences (Penzberg, Germany). Glutathione (GSH, A005-1-2), superoxide dismutase (SOD, A001-3-2), and malondialdehyde (MDA, A003-1-2) assay kits were purchased from Jiancheng Biological Engineering (Nanjing, Jiangsu, China). Real-time polymerase chain reaction (PCR) reagents were purchased from Thermo Fisher (Waltham, Massachusetts, USA). METH was purchased from National Institutes for Food and Drug Control (Beijing, China). FIHMI was provided by Professor Sun from Soochow University and prepared in dimethyl sulfoxide (DMSO).
Cell culture and drug treatments
661W cells were cultured in RPMI1640 medium supplemented with 10% fetal bovine serum and 1% streptomycin/penicillin and incubated at 37°C in a humidified atmosphere of 5% CO2 and 95% air. 661W cells were exposed to METH for 24 h. FIHMI was added to the culture medium at determined concentrations 6 h before METH treatment.
Cell viability assay
Cell viability was evaluated by MTT assay. Briefly, after treatment, 661W cells were seeded in the 96-well plate. MTT was added in a volume of 10 μL to each well (0.5 mg/mL final concentration) and incubated for 4 h. The MTT-containing medium was replaced by 200 μL DMSO, and the plates were incubated for 10 min at 37°C. The absorbance of each well was measured at 570 nm using an enzyme-linked immunosorbent assay (ELISA) microplate reader (Perkin Elmer, Waltham, Massachusetts, USA).
MMP measurement
Mitochondrial membrane potential (MMP) was measured by the fluorescent dye JC-1. Briefly, cells were washed and incubated with JC-1 in the dark for 15 min at 37°C. After rinsing twice, the intensity of fluorescence was measured by the plate reader (BioTek, Winooski, Vermont, USA) at the excitation of 490 nm and emission of 530/590 nm, respectively.
Cytochrome c assay
Cells were fractionated after treatment and the cytochrome c level was measured by the kit following manufacturer’s instructions.
Caspase activity measurement
Activities of caspase 3/9 were measured according to the manufacture’s protocol. Briefly, 100 µL of reagent for specific caspase was added into each well, and the plate was then incubated at room temperature for 3 h. Luminescence of samples was measured using a microplate reader (Perkin Elmer, Waltham, Massachusetts, USA).
DNA fragmentation measurement
Cells were lysed after treatment, and DNA fragmentation was measured by Cell Death Detection ELISAplus kit following manufacturer’s instructions.
SOD activity, MDA, and GSH level
SOD activity, MDA, and GSH levels were measured by the assay kits according to the manufacturer’s protocol. Briefly, after washing, 661W cells were homogenized, and supernatant was used to measure SOD, MDA, and GSH.
Measurement of ROS
Intracellular ROS was measured by the fluoroprobe DCF-DA. 661W cells were incubated with DCF-DA for 30 min in the dark. After washing twice, the fluorescence intensity was measured at 488/525 nm using a fluorescence microplate reader (BioTek, Winooski, Vermont, USA).
Real-time PCR
Total RNA was extracted with 1 mL TRIzol reagent (Thermo Scientific, Waltham, Massachusetts, USA). cDNA was generated with 0.5 µg total RNA using iScript™ Reverse Transcription Supermix kit (BioRad, Hercules, California, USA) following manufacturer’s instructions. Real-time PCR was run with SYBR Green Supermix reagents (Thermo Scientific). The relative mRNA expression of interest in different samples was quantified by comparative Ct value method. The mRNA expression was normalized to the housekeeping gene β-actin. The gene-specific primer sequences are the following. For Beclin-1, forward: ggccaataagatgggtctga; reverse: gctgcacacagtccagaaaa, NCBI reference: NM_019584.4; for β-actin, forward: tgttaccaactgggacgaca; and reverse: ggggtgttgaaggtctcaaa, NCBI reference: NM_007393.5.
Western blot
Protein was extracted with radioimmunoprecipitation assay protein lysate buffer, supplemented with protease inhibitors (Roche, Basel, Switzerland) and phosphatase inhibitors (Thermo Scientific). After boiling, samples with 40 μg protein were run with the 12% (v/v) sodium dodecyl sulfate–polyacrylamide gel and then electroblotted from gel to the polyvinylidene difluoride membrane. After blocking with phosphate-buffered saline containing 1% bovine serum albumin for 1 h at room temperature, the membrane was incubated with primary antibodies (anti-Bcl-2, rabbit pAb ab196495, 1:1500; anti-Bax rabbit pAb ab199677, 1:1000; anti-LC3B rabbit pAb ab48394, 1:1000; anti- nicotinamide adenine dinucleotide phosphate (NADPH) oxidase 4 rabbit pAb ab154244, 1:1000; anti-nuclear factor (erythroid-derived 2)-like 2 (Nrf2) rabbit pAb ab31163, 1:1000; and anti-β-actin, rabbit pAb, ab1801, 1:3000) overnight at 4°C, followed by incubating with the horseradish peroxidase-conjugated secondary antibody (goat anti-rabbit IgG H&L HRP, ab6721, 1:3000) for 1 h at room temperature. Membrane was then washed three times and visualized by the enhanced chemiluminescence system.
Statistical analysis
Data were expressed as mean ± SD and analyzed with GraphPad Prism 6 software (GraphPad Software Inc., San Diego, California, USA). Data were compared by one-way analysis of variance, followed by Tukey’s post hoc analysis. The value of p < 0.05 was considered statistically significant. All experiments were replicated three times.
Results
Effects of FIHMI on the viability of METH-treated 661W cells
Effects of METH on 661W cell viability were examined by MTT assay. Our data showed that METH decreased the viability of 661W cells in a dose-dependent manner. We further investigated whether FIHMI protected 661W cells against METH-induced toxicity. Results showed 6 h pretreatment of FIHMI significantly increased cell viability. Moreover, FIHMI treatment alone showed no obvious effects on 661W cell viability (Figure 1).
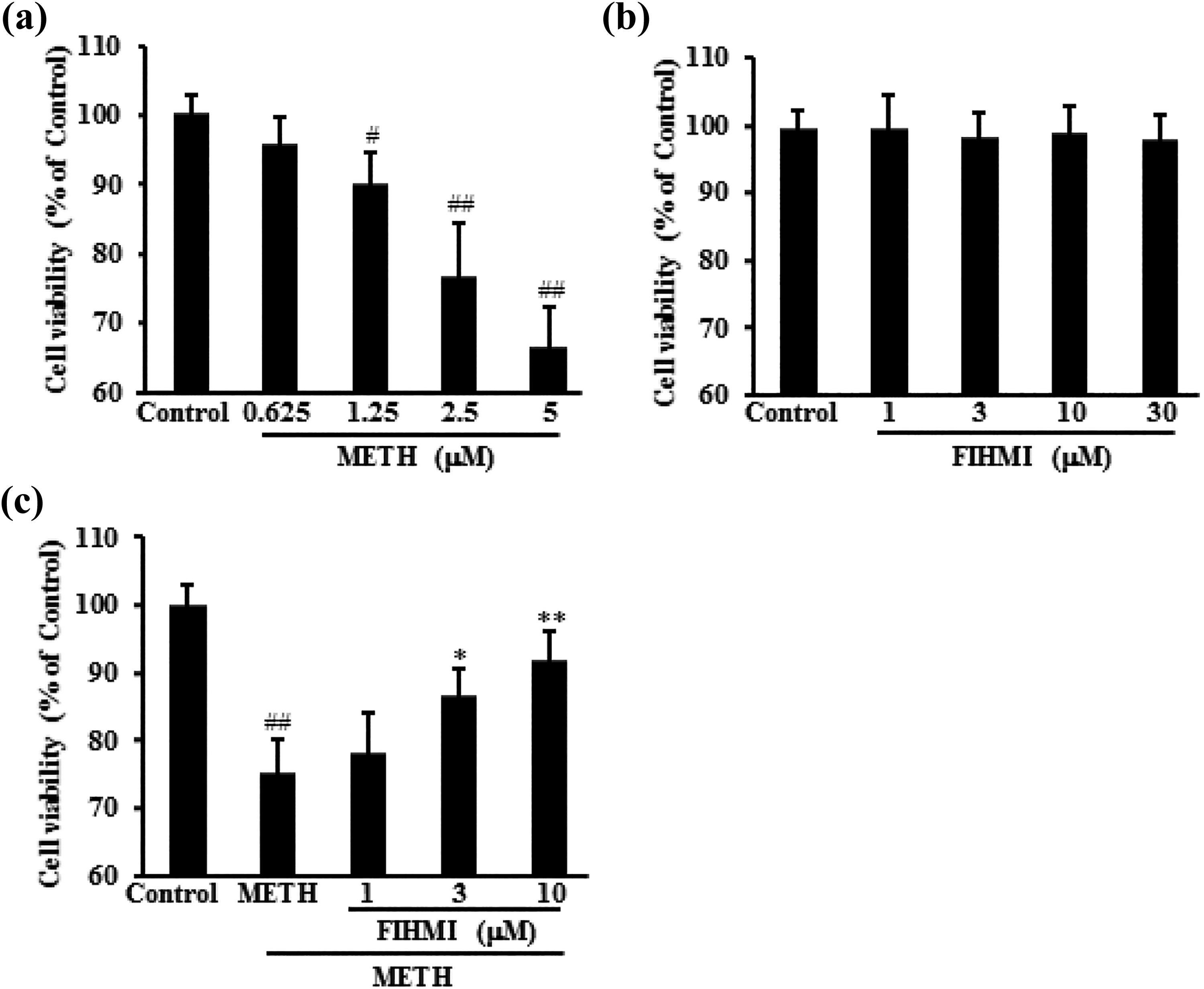
FIHMI protected 661W cells against METH-induced cell toxicity. (a) Effects of METH on cell viability. 661W cells were treated with METH and the cell viability was measured by MTT assay. (b) Effects of FIHMI on cell viability. 661W cells were treated with FIHMI and the cell viability was measured by MTT assay. (c) FIHMI pretreatment decreased cell toxicity of 661W cells induced by METH. Cells were treated with 2.5 µM METH for 24 h in the absence or presence of FIHMI 6 h prior to METH treatment. Data were expressed as mean ± SD. #p < 0.05, ##p < 0.01 versus control; *p < 0.05, **p < 0.01 versus METH alone. FIHMI: 6-formyl-5-isopropyl-3-hydroxymethyl-7-methyl-1H-indene; METH: methamphetamine; MTT: 3-(4,5-dimethylthiazol)-2,5-diphenyltetrazolium bromide.
FIHMI treatment attenuated 661W cell damage caused by METH
METH treatment caused toxicity to 661W cells, however, pretreatment with FIHMI increased MMP (Figure 2(a)), decreased cytochrome c release (Figure 2(b)), DNA fragmentation (Figure 2(c)) as well as caspase activities (Figure 2(d)), and changed the expression of apoptosis-related protein (Figure 2(e)).

FIHMI protected 661W cells against METH-induced cell damage. 661W cells were treated with FIHMI for 6 h followed by METH treatment for 24 h. Cells were collected to measure the cytotoxicity: (a) mitochondrial membrane potential, (b) cytochrome c release, (c) DNA damage, and (d) caspase activities. (e) The apoptosis-related proteins were also detected by Western blot. Data were expressed as mean ± SD. ##p < 0.01 versus control; *p < 0.05, **p < 0.01 versus METH alone. FIHMI: 6-formyl-5-isopropyl-3-hydroxymethyl-7-methyl-1H-indene; METH: methamphetamine.
FIHMI treatment decreased autophagy in 661W cell caused by METH
Effects of FIHMI on 661W cell autophagy induced by METH were explored by examining the mRNA expression of Beclin-1 and LC3B protein level. Results showed that pretreatment with FIHMI decreased Beclin-1 mRNA expression and LC3B protein level in METH-treated 661W cells (Figure 3).

Effects of FIHMI on autophagy in METH-treated 661W cells. 661W cells were treated with FIHMI for 6 h, followed by METH treatment for 24 h. Cells were collected to measure the mRNA and protein expression. (a) Beclin-1 mRNA expression. (b) LC3B protein expression. Data were expressed as mean ± SD. ##p < 0.01 versus control; *p < 0.05, **p < 0.01 versus METH alone. FIHMI: 6-formyl-5-isopropyl-3-hydroxymethyl-7-methyl-1H-indene; METH:=methamphetamine.
FIHMI treatment decreased the oxidative stress in 661W cells caused by METH
Oxidative stress significantly increased after METH treatment. However, pretreatment with FIHMI significantly reduced the production of ROS and MDA and increased GSH levels and SOD activities, together with increasing the protein expression of Nrf2 and decreasing the protein expression of NADPH oxidase 4, indicating FIHMI treatment effectively decreased the oxidative stress induced by METH (Figure 4).

FIHMI inhibited oxidative stress in 661W cells induced by METH. 661W cells were treated with FIHMI 6 h, followed by METH treatment for 24 h. Cells were collected to measure the oxidative stress markers: (a) intracellular ROS level, (b) MDA content, (c) GSH level, (d) SOD activity, and (e) NADPH oxidase 4 and Nrf2 protein expression. Data were expressed as mean ± SD. ##p < 0.01 versus control; *p < 0.05, **p < 0.01 versus METH alone. FIHMI: 6-formyl-5-isopropyl-3-hydroxymethyl-7-methyl-1H-indene; METH: methamphetamine; ROS: reactive oxygen species; MDA: malondialdehyde; GSH: glutathione; SOD: superoxide dismutase; NADPH: nicotinamide adenine dinucleotide phosphate.
Discussion
METH exposure can cause cell damage to the dopaminergic axon terminals, which have been associated with oxidative stress. 14,15 METH is also known as a neurotoxic model, which is involved in inflammatory–neurological processes and brain dysfunction due to abuse of the drug. Some studies showed oxidative stress, apoptosis, necrosis, and calcium deregulation are associated with METH-induced toxicity. 16 –18 The antioxidant activities of FIHMI was suggested as a plausible mechanism for its protective potential. Therefore, this study examined the protective effects of FIHMI on METH-induced toxicity in photoreceptor cells. Results demonstrated that METH significantly reduced cell viability comparing with the control group, while cell viability could be increased when 661W cells were pre-treated with FIHMI.
Apoptosis involves in multiple pathways driving to a change in the structure of biomolecules, including the modification of the membrane structure and consequently, the permeability of the membrane to the different materials. 19 Formation and accumulation of ROS in the mitochondria increases mitochondrial membrane permeability and decreases mitochondrial membrane potential. Cytochrome c, a proapoptotic protein, flows from the damaged mitochondrial membrane into the cytoplasm. Leakage of cytochrome c activates caspases and nuclear fragmentation to initiate apoptosis. 20 In this study, METH treatment caused cell damage, evidenced by MMP decrease, cytochrome c release, and increased activities of caspase 3/9. Antiapoptotic protein expression decreased, but proapoptotic protein expression increased. However, the above effects were largely reversed by FIHMI pretreatment.
Autophagy plays an essential role in the growth, homeostasis, and cell differentiation, which can be activated upon physiological stresses such as cell protection response to intracellular stresses, toxic metabolites, and intracellular pathogens. 21,22 In the lack of peculiar control, autophagy may stimulate apoptosis and degrade cells by self-digestion and eventually leading to death. METH induces autophagy as a survival response to the death of apoptotic endothelial cells through the kappa opioid receptor. Prolonged exposure to METH is flushing autophagy and dooming the cell to apoptosis. 23 In this study, Beclin-1 mRNA and LC3B protein expression was examined. Results showed exposing 661W cells to METH caused a significant increase in the expression of Beclin-1 and LC3B. However, the increase can be reversed by FIHMI pre-treatment.
Excessive accumulation of ROS can result in oxidative stress and high oxidative stress causes different cell death patterns, including apoptosis, autophagy, and necrosis. 24,25 Improved levels of ROS magnifies oxidative stress response and excessive generation of ROS stimulates hypoxia-inducible factor and p53. MDA is a widely used marker for oxidative damage and resultant thiobarbituric acid reactive substances, which are proportional to oxidative stress and lipid peroxidation. 26 Then, these factors induce the transcription of target genes such as LC3, BNIP3, and p62. 27,28 Nrf2 is a master regulator of antioxidant and detoxificant genes with cytoprotective function. 29 Activation of Nrf2 may protect cells from apoptosis, through binding with antioxidant response element and inducing cytoprotective target protein expression. 30 In this study, FIHMI-induced Nrf2 activation was observed, which further triggered expression of antioxidant genes to restore oxidative homeostasis, showed by reduced levels of MDA and ROS, and increased SOD activity as well as GSH level.
In conclusion, our study demonstrated that treatment of 661W cells with METH resulted in apoptosis and autophagy. FIHMI was effective in protecting 661W cells against METH-induced toxicity providing the scientific rationale to develop FIHMI as a therapeutic agent against METH-induced photoreceptor cell damage.
Footnotes
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.