
Abstract
Triptolide is a major active ingredient isolated from the traditional Chinese herb Tripterygium wilfordii Hook F. However, its use in clinical practice is limited due to its severe hepatotoxicity. Autophagy, a highly conserved intracellular process, is essential for maintaining cytoplasmic homeostasis. Considering that abnormalities in autophagy are closely associated with drug-mediated hepatotoxicity, we applied human normal liver HL7702 cells to elucidate the roles of autophagy in triptolide-induced hepatotoxicity. Our study revealed that triptolide was cytotoxic to HL7702 cells. It markedly increased autophagosome formation and expression of autophagy-related proteins, namely Beclin1 and microtubule-associated protein 1 light chain 3II, and induced oxidative stress. These proautophagic effects were counteracted by pretreatment with N-acetylcysteine, a reactive oxygen species scavenger. Moreover, the pharmacological suppression of autophagy further exacerbated triptolide-elicited decrease in cell viability, increase in lactate dehydrogenase leakage, and activation of apoptosis proteases (caspase 3 and caspase 9). Our findings suggest that triptolide-induced oxidative stress consequently enhances autophagic activity, and autophagy is a cytoprotective mechanism against triptolide-induced cytotoxicity in HL7702 cells.
Introduction
Triptolide, a diterpene triepoxide, is the primary active constituent extracted from Tripterygium wilfordii Hook F. It possesses several pharmacological activities, including immunosuppressive, anti-inflammatory, and antineoplastic effects. 1 However, its clinical applications are limited due to its severe hepatotoxicity. In particular, triptolide primarily accumulates in the liver, making the liver a major target organ for the toxic effects of triptolide. 2,3 Although triptolide hepatotoxicity has been extensively investigated, its definitive mechanisms remain poorly characterized.
Autophagy, a catabolic process involving delivery of cytoplasmic organelles and long-lived proteins for lysosomal degradation and recycling, is necessary for maintaining cellular homeostasis. Its dysfunction contributes to a wide variety of human pathologies, including neurodegeneration, cancer, aging, and infectious diseases. 4 Accumulating evidence shows that autophagy is intimately implicated in drug-induced hepatotoxicity. For example, acetaminophen-activated autophagy removes damaged mitochondria and protects against liver toxicity. 5 In addition, efavirenz treatment increases autophagic activity in human hepatic cells, and its toxic effects are aggravated by an autophagy inhibitor. 6 Similarly, dasatinib-induced autophagy is accompanied by the development of liver damage, and suppression of autophagy exacerbates dasatinib-induced hepatotoxicity. 7
Emerging evidence has proven that the autophagic pathway is one of the action targets of triptolide in in vitro and in vivo models. 8 Triptolide induces cell death in leukemia cell lines by stimulating autophagy, 9 and autophagic activity increases in triptolide-treated orthotopic tumor samples of pancreatic cancer cell in nude mice. 10 In neuronal cells, triptolide promotes α-synuclein degradation by modulating autophagy and protects against α-synuclein-induced cytotoxicity. 11 Autophagy also plays a critical role in triptolide-induced cardiotoxicity. 12 Triptolide exposure impairs the mitochondrial function of hepatocytes, abnormally increases reactive oxygen species (ROS) level, decreases antioxidant enzyme activity, and subsequently causes oxidative stress in liver tissues. 13,14 In addition, oxidative stress is a potent contributor to autophagy initiation and is invariably involved in the outcome of autophagy. 12 Thus, autophagy may be altered in triptolide-treated hepatic cells and participate in hepatotoxicity. Additionally, apoptosis participates in triptolide-induced hepatotoxicity. Triptolide-triggered apoptosis is mediated through the mitochondrial pathway and involves the caspase family. Triptolide induces apoptosis in human liver cells by causing mitochondrial impairment, promoting the release of cytochrome c from the mitochondria to the cytosol, and upregulating the expression levels of Bax, p53, caspase 3, and caspase 9 concurrently with the protein levels of Bcl-2. 2,13 Meanwhile, autophagy and triptolide-induced apoptosis are intimately interrelated. For example, triptolide-mediated apoptotic cell death is attenuated in cardiomyocytes by upregulating autophagy 12 but is accelerated in prostate cancer cells by suppressing autophagy. 15 Hence, autophagy likely participates in regulating triptolide-mediated apoptosis in hepatocytes.
In this study, human normal liver HL7702 cells were employed to evaluate whether triptolide modulates autophagic activity and to delineate the potential roles of autophagy in triptolide-mediated hepatotoxicity. This study may provide insights into the mechanism underlying triptolide-induced hepatotoxicity. It may also guide the development of strategies for alleviating triptolide-induced hepatotoxicity through autophagy modulation.
Materials and Methods
Reagents
Triptolide (>99% purity) was obtained from Winherb Medical Science Co (Shanghai, China). Triptolide was dissolved in dimethyl sulfoxide, and stock solutions were stored at −20°C.Cell Counting Kit 8 (CCK8) was bought from Dojindo Molecular Technologies, Inc (Kumamoto, Japan). Rabbit anti-light chain 3 (LC3) polyclonal antibody, goat anti-actin polyclonal antibody, horseradish peroxidase–conjugated goat antirabbit immunoglobulin G (IgG), goat antimouse IgG, and donkey antigoat IgG were obtained from Beyotime Biotechnology (Haimen, China). Mouse anti-Beclin1 monoclonal antibody, bafilomycin A1, and chloroquine (CQ) were obtained from Solarbio Science & Technology Co, Ltd (Beijing, China). Dichlorodihydrofluorescein diacetate (DCFH-DA) and N-acetylcysteine (NAC) were ordered from Sigma-Aldrich (St Louis, Missouri). Superoxide dismutase (SOD), glutathione (GSH), malondialdehyde (MDA), lactate dehydrogenase (LDH), caspase 3, and caspase 9 assay kits were purchased from Jiancheng Bioengineering Institute (Nanjing, China).
Cell Culture and Drug Treatment
HL7702 cells were grown in Dulbecco’s modified Eagle’s medium (Gibco, Grand Island, Nebraska) supplemented with 10% fetal bovine serum (Invitrogen, Carlsbad, California) and penicillin/streptomycin at 37°C in a humidified 5% CO2 incubator. Cells were treated with triptolide, bafilomycin A1, CQ, or NAC as described in individual experiments.
Assessment of Cell Viability
Cell viability was obtained using CCK8 kit according to the manufacturer’s instructions. 16 All experiments were performed in triplicate.
Lactate Dehydrogenase Leakage Assay
HL7702 cells were seeded in 96-well plates at a density of 1 × 104 cells per well and treated with different concentrations of triptolide for 24 hours. Cell damage was then assessed by determining LDH release from cells by using an LDH detection kit in accordance with the manufacturer’s protocol. 14
Quantitative Real-Time Polymerase Chain Reaction Analysis
HL7702 cells were treated with different doses of triptolide for the desired duration. Total RNA was extracted using TRIzol reagent (Invitrogen), and complementary DNA was prepared from total RNA by using RevertAid Premium Reverse Transcriptase (Thermo Scientific, Waltham, Massachusetts) in accordance with the manufacturer’s instructions. The sequences of the real-time polymerase chain reaction primers for Beclin1 were designed as 5′-AAACCAGATGCGTTATGCCC-3′ and 5′-CAGCCTGAAGTTATTGATTGTGC-3′; for actin, the sequences were 5′-TAGTTGCGTTACACCCTTTCTTG-3′ and 5′-TCACCTTCACC GTTCCAGTTT-3′. Polymerase chain reaction amplification was subsequently performed with a StepOne system (ABI, Foster City, California). The messenger RNA (mRNA) level of each gene was normalized to that of actin by using the ΔΔCT method.
Measurement of Intracellular ROS
The fluorescent probe DCFH-DA was used to detect intracellular ROS levels. Briefly, HL7702 cells were seeded in 96-well black plates. After reaching 60% to 70% confluence, they were exposed to triptolide for 24 hours. After incubation, they were washed with phosphate-buffered saline (pH 7.4) followed by staining with DCFH-DA at 37°C for 30 minutes. The fluorescence intensity of the cells was examined using a Monochromator-Based Multimode Microplate Reader (Infinite M1000; Tecan, Männedorf, Switzerland) with excitation wavelength at 488 nm and emission wavelength at 525 nm.
Measurement of SOD, GSH, and MDA
The activities of the antioxidant enzyme SOD in HL7702 cells were determined by using an assay kit. The GSH or MDA contents were also measured by using commercial kits in accordance with the manufacturer’s instructions. 14
Cell Lysate Preparation and Western Blot Analysis
Cells were harvested and lysed in 1 × radioimmunoprecipitation assay buffer (1% NP40, 50 mM Tris–HCl [pH 7.4], 0.25% sodium deoxycholate, 150 mM NaCl, and 1 mM EDTA) containing a protease inhibitor mixture (P8340; Sigma-Aldrich). Lysates were cleared through centrifugation at 1,000g for 10 minutes at 4°C after 20 minutes of incubation on ice. Supernatants were mixed with loading buffer and boiled at 95°C for 5 minutes. Cell lysates were subjected to sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE). After SDS-PAGE, proteins were transferred onto nitrocellulose membranes in 25 mM Tris–HCl, 192 mM glycine, and 20% (vol/vol) methanol. Membranes were blocked with 5% milk in Tris-buffered saline with 0.1% Tween 20 (TBST) at room temperature following incubation with the indicated primary antibodies. After overnight incubation with the primary antibodies (1:1,000 dilution), membranes were washed with TBST and probed with the indicated secondary antibodies (1:5,000 dilution) at room temperature. The membranes were then washed with TBST prior to the visualization of protein bands by using the ECL system (Beyotime Biotechnology, Haimen, China). Signal intensity was quantified by ImageJ (National Institutes of Health, Bethesda, Maryland).
Transmission Electron Microscopy for Autophagosomes
To directly characterize the ultrastructure of autophagic vesicles, HL7702 cells were collected and fixed overnight with 2.5% glutaraldehyde at 4°C, postfixed in 1% osmium tetroxide, dehydrated through serial dilution in ethanol, and embedded in epoxy resin. Ultrathin sections were observed under an Hitachi 7800 transmission electron microscope (Tokyo, Japan).
Detection of Caspase 3 and Caspase 9 Activities
Caspase 3 and caspase 9 activities were quantified in accordance with the manufacturer’s experimental instructions. All experiments were performed in triplicate. 17
Statistical Analysis
Data are expressed as the mean ± standard error of the mean. For comparison of 2 groups, the statistical difference is determined by t test; P value <0.05 is considered statistically significant in all tests.
Results
Cytotoxic Effects of Triptolide on HL7702 Cells
HL7702 cells were treated with 0.05, 0.1, or 0.2 μM triptolide for 24 hours or with 0.1 μM triptolide for 12, 24, and 48 hours to observe the cytotoxic effects of triptolide. Cell viability was then assessed. As shown in Figure 1A, the cell viability of group incubated with triptolide reduced in a dose- and time-dependent manner relative to that of the control group. The LDH leakage was measured to determine the cell membrane integrity. As shown in Figure 1B, triptolide increased LDH release by promoting cell injury. These data suggest that triptolide treatment exerts cytotoxic effects on HL7702 cells.
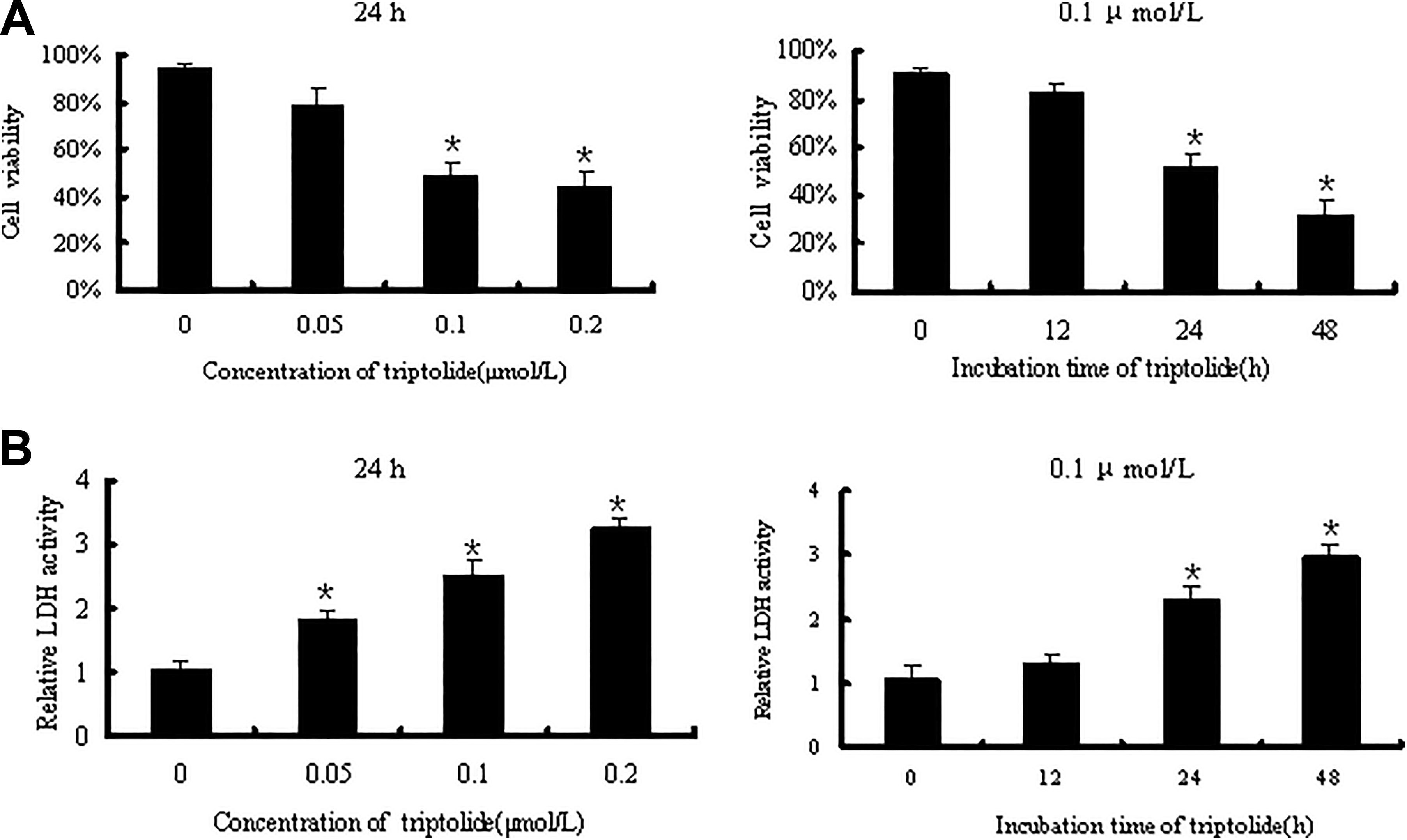
Concentration- and time-dependent cell viability (A) and LDH leakage (B) of HL7702 cells with triptolide treatments. When comparing the control, obvious differences can be seen. The LDH leakage is expressed as the folds of the control values. Data are represented as the mean ± SEM from 3 independent experiments in triplicate; *P < 0.05 versus control group. LDH indicates lactate dehydrogenase; SEM, standard error of the mean.
Triptolide Induces Autophagy in HL7702 Cells
HL7702 cells were exposed to 0.05, 0.1, or 0.2 μM triptolide or treated with 0.1 μM triptolide for 12, 24, or 48 hours to investigate whether triptolide incubation induces autophagy in HL7702 cells. Then, the mRNA expression levels of Beclin1, a protein that is essential for autophagy initiation, was examined. As shown in Figure 2A, triptolide enhanced the expression of Beclin1 mRNA in a concentration- and time-dependent manner. Moreover, the results of Western blot analysis confirm that triptolide treatment drastically increased Beclin1 expression. The expression of autophagosome membrane-bound protein–microtubule-associated protein 1 LC3II, which localizes in particular on autophagosome membranes and is a hallmark of autophagy, 18 was measured. The LC3II content of HL7702 cells increased in a dose- and time-dependent manner (Figure 2B and C). Autophagic flux was measured by using a combination of the autophagy inhibitor bafilomycin A1, which inhibits autophagosome–lysosome fusion, 19 to clarify whether the triptolide-induced autophagy was due to increased autophagosome formation or decreased degradation given that LC3II itself is an autophagy substrate. The further accumulation of LC3II in the presence of bafilomycin A1 is suggestive of the enhancement of autophagic flux. 20 Indeed, triptolide-induced LC3II accumulation was further elevated under treatment with bafilomycin A1 (P < 0.05; Figure 2D). Finally, to further determine the induction of autophagy by triptolide, the existence of autophagosomes was determined through transmission electron microscopy. As shown in Figure 2E, numerous autophagic vesicles characterized by membrane-bound vacuoles containing organelles and cellular fragments were observed in the cytoplasm of triptolide-treated HL7702 cells.
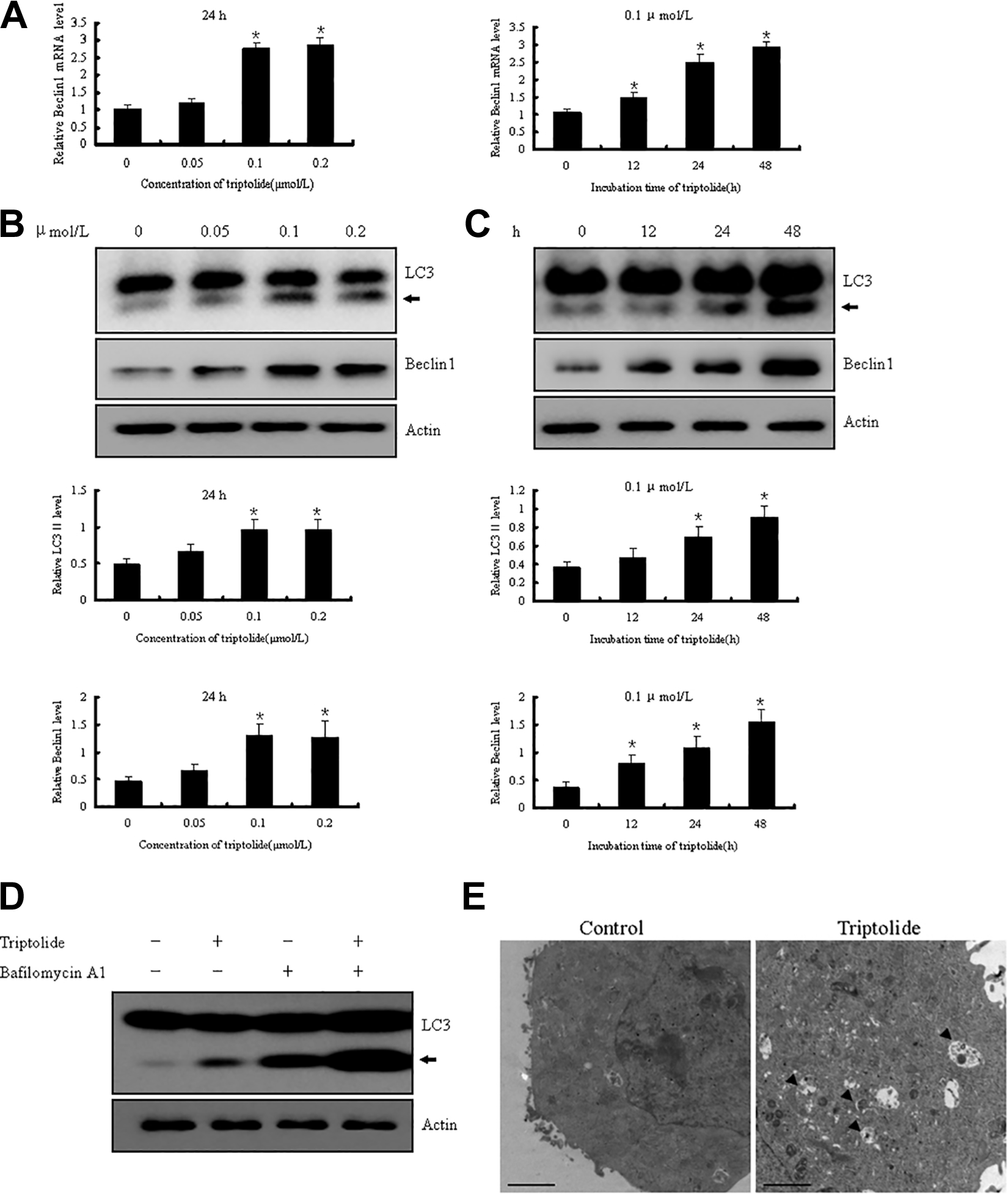
Concentration- and time-dependent Beclin1 mRNA (A), and LC3II and Beclin1 protein expression (B and C) in HL7702 cells with triptolide treatments. Obvious increases in Beclin1 mRNA, LC3II, and Beclin1 protein expression are observed comparing the control. Representative blots are shown. The arrowhead indicates the LC3II protein. The signal intensities were normalized with actin and expressed as the mean ± SEM (n = 3); *P < 0.05 versus control group. D, The LC3II expression of HL7702 cells with triptolide 0.1 μM treatment in the presence or absence of 0.1 μM bafilomycin A1 for 24 hours in DMEM containing 10% FBS. Bafilomycin A1 inhibits autophagosome–lysosome fusion, which enhanced triptolide-induced LC3II expression. A representative blot from 3 independent experiments is shown. E, Transmission electron microscopy analysis for HL7702 cells with triptolide treatment (0.1 μM, 24 hours) in DMEM containing 10% FBS (scale bar = 2.0 μm). Arrows indicate autophagic vacuoles. DMEM indicates Dulbecco’s modified Eagle’s medium; FBS, fetal bovine serum; LC3II, microtubule-associated protein 1 light chain 3II; mRNA, messenger RNA; SEM, standard error of the mean.
Triptolide Induces Oxidative Stress in HL7702 Cells
HL7702 cells were exposed to various concentrations of triptolide for 24 hours to determine the effects of triptolide on oxidative stress markers, including the generation of ROS, the activity of the intracellular antioxidant SOD, the content of GSH, and the level of the lipid peroxidation product MDA. As shown in Figure 3A, treatment with 0.1 and 0.2 μM triptolide significantly increased intracellular ROS production (P < 0.05). Meanwhile, triptolide reduced SOD activities and GSH content in a dose-dependent manner (Figure 3B). Accordingly, the MDA levels of cells exposed to 0.1or 0.2 μM triptolide for 24 hours significantly increased compared with those of the control (P < 0.05; Figure 3C). These results imply that triptolide induces oxidative stress in HL7702 cells.
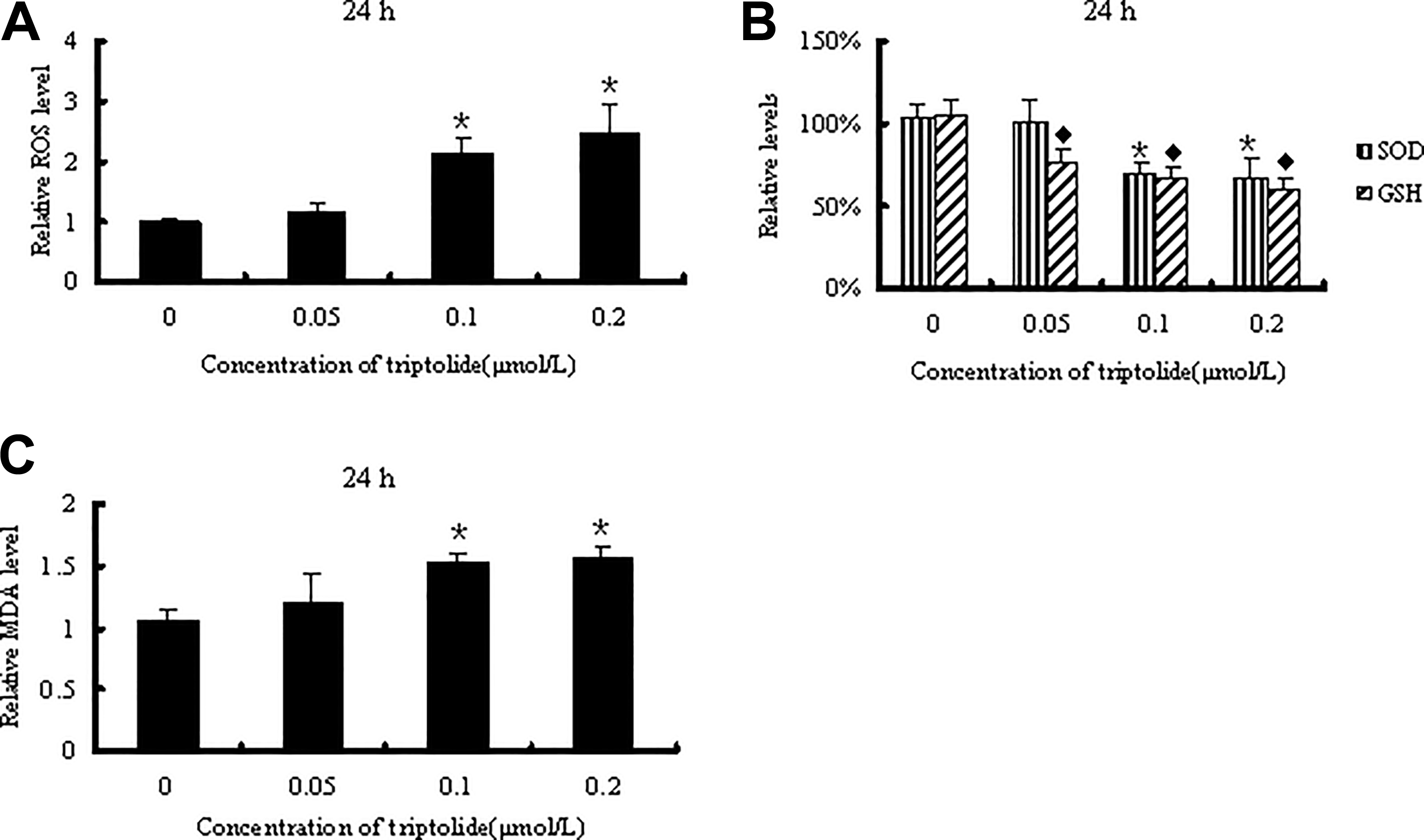
Intracellular ROS (A), antioxidant SOD activity, GSH content (B), and MDA levels (C) in HL7702 cells following the indicated triptolide treatment in DMEM containing 10% FBS. Obvious differences can be seen when comparing the control. Data are shown as the folds of the control and represented as the mean ± SEM from 3 independent experiments in triplicate; *P < 0.05 versus control group. DMEM indicates Dulbecco’s modified Eagle’s medium; FBS, fetal bovine serum; GSH, glutathione; MDA, malondialdehyde; ROS, reactive oxygen species; SEM, standard error of the mean; SOD, superoxide dismutase.
Oxidative Stress Involved in Triptolide-Induced Autophagy
Given that triptolide led to oxidative stress, a potent autophagy stimulus, 21 NAC, a general ROS scavenger, was applied to block intracellular ROS generation to evaluate the potential importance of oxidative stress in triptolide-induced autophagy. Pretreating HL7702 cells with NAC for 4 hours prior to triptolide treatment significantly blocked ROS production (P < 0.05; Figure 4A). The enhancement in LC3II expression caused by triptolide was markedly attenuated in the presence of NAC (P < 0.05; Figure 4B). These results indicate that triptolide-induced oxidative stress regulates autophagy activation.
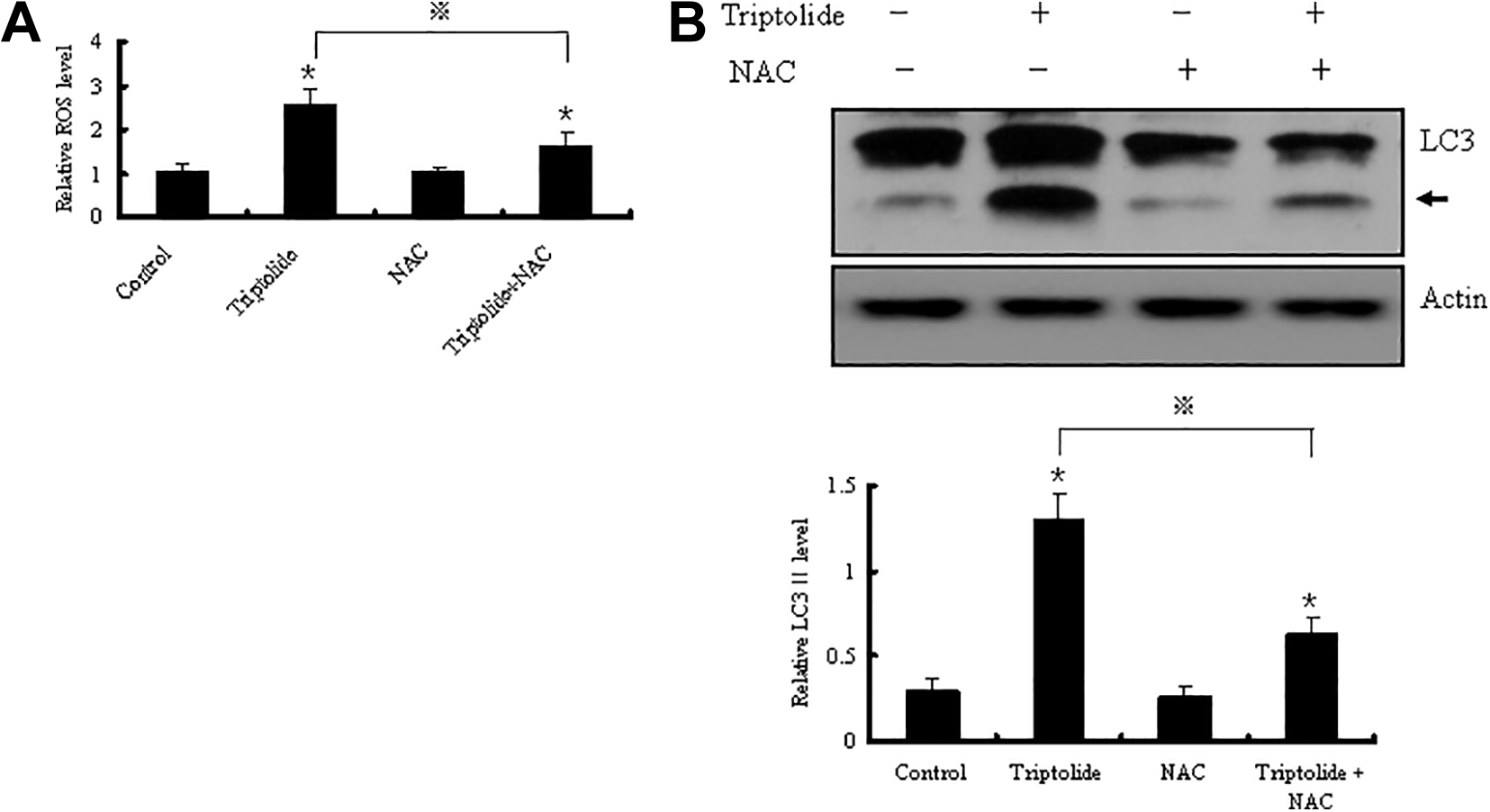
Intracellular ROS (A) and LC3II protein expression (B) in HL7702 cells induced by triptolide (0.1 μM, 20 hours) with or without NAC pretreatment (10 mM, 4 hours) in DMEM containing 10% FBS; NAC is the known ROS scavenger. It shows that NAC inhibited triptolide-induced enhancement of ROS and LC3II. A representative blot for LC3II is shown, and the signal intensities were normalized with actin. Data are represented as the mean ± SEM (n = 3); *,⋄P < 0.05 versus control group; ※P < 0.05 versus triptolide-treated group. DMEM indicates Dulbecco’s modified Eagle’s medium; FBS, fetal bovine serum; LC3II, microtubule-associated protein 1 light chain 3II; NAC, N-acetylcysteine; ROS, reactive oxygen species; SEM, standard error of the mean.
Autophagy Plays Protective Roles in Triptolide-Mediated Cytotoxicity in HL7702 Cells
Chloroquine was applied to disrupt the late stage of the autophagic pathway, 22 and cell viability, LDH leakage, and caspase activities were assessed to address the role of autophagy in triptolide-induced cytotoxicity in HL7702 cells. As shown in Figure 5A, cotreatment with CQ dramatically decreased the viability of HL7702 cells in the presence of triptolide (P < .05). In the meantime, LDH leakage in the triptolide and CQ treatment group was elevated relative to that in the triptolide treatment group (Figure 5B). Moreover, triptolide treatment increased the activities of apoptosis markers caspase 3 and caspase 9 simultaneously (P < .05). The significant upregulation of caspase 3 and caspase 9 activities (P < 0.05) reflects the stronger proapoptotic effect of triptolide in combination with CQ relative to that of triptolide alone (Figure 5C).
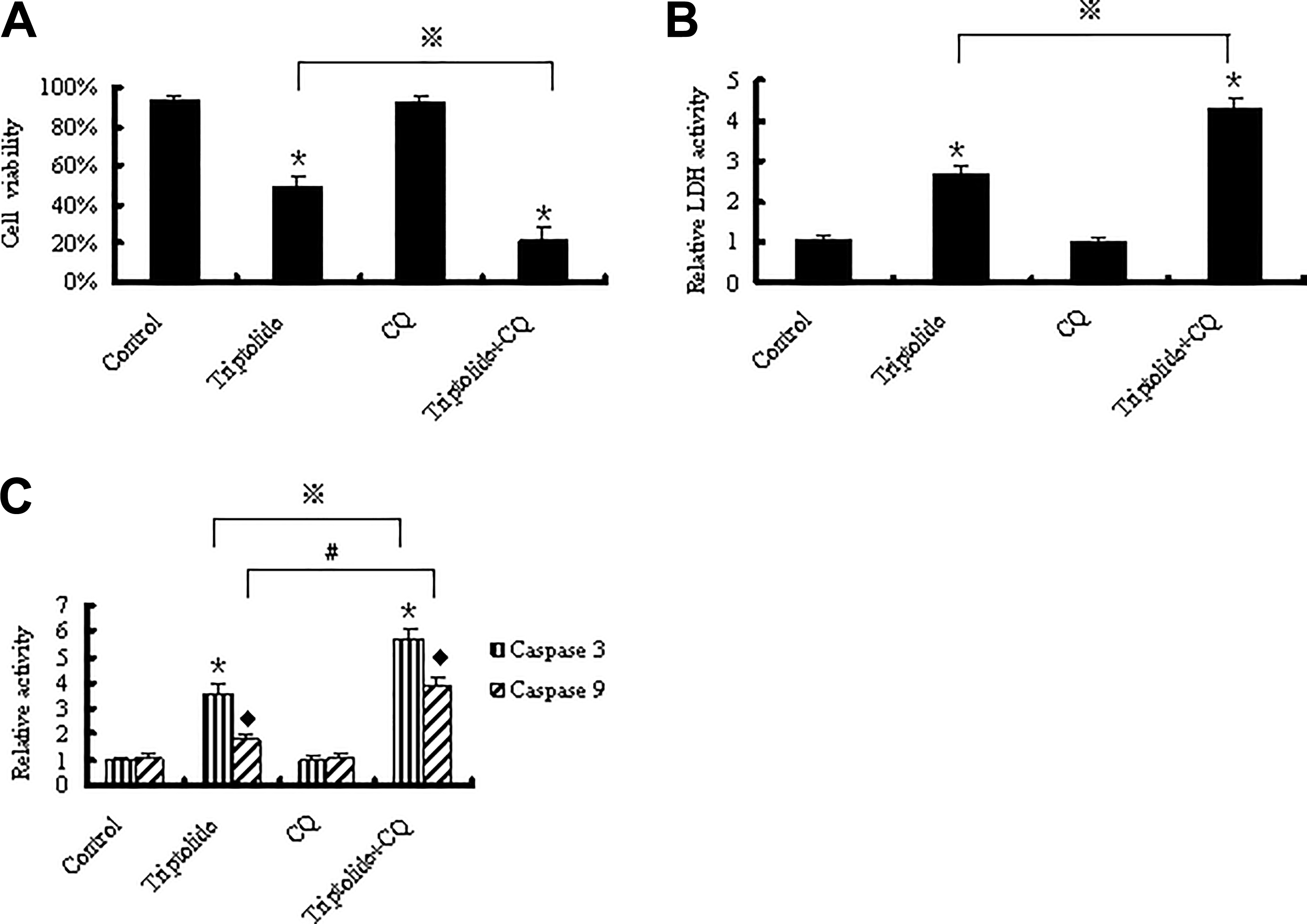
Cell viability (A), LDH leakage (B), and caspase 3 and caspase 9 activities (C) of HL7702 cells with triptolide 0.1 μM treatment in the presence or absence of 10 μM CQ for 24 hours. Through disruption of autophagic pathway, CQ cotreatment enhanced triptolide-induced decrease in cell viability, elevation of LDH leakage, and caspase 3 and caspase 9 activities. Lactate dehydrogenase leakage, caspase 3, and caspase 9 are expressed as the folds of the control values and represented as the mean ± SEM from 3 independent experiments in triplicate; *,⋄P < 0.05 versus control group; ※, #P < 0.05 versus triptolide-treated group. CQ indicates chloroquine; LDH, lactate dehydrogenase; SEM, standard error of the mean.
Discussion
Human normal liver HL7702 cells showed time- and dose-dependent reductions in viability and enhanced LDH leakage upon triptolide exposure. These results indicate that triptolide has a toxic action against hepatic cells. Hepatotoxicity is one of the most prevalent triptolide-related side effects. Triptolide reduces viability and induces apoptosis in cultured hepatocytes. In vivo experiments have shown that triptolide administration results in acute hepatic injury and drastically elevates levels of serum aspartate aminotransferase and alanine transaminase. 2,3,14,23 Various mechanisms have been proposed to be responsible for the hepatotoxic effects of triptolide. 24 Autophagy is involved in the hepatotoxicity of several drugs. 5 -7 However, the role of autophagy in triptolide-induced hepatotoxicity remains unclear. Herein, we observed that triptolide increased the expression levels of the autophagy-related proteins Beclin1 and LC3II, promoted autophagosome formation, and activated autophagic flux. These phenomena suggest that triptolide upregulates autophagic activity in HL7702 cells. Moreover, the antitumor, neuroprotective, cardiac toxic, and podocyte protective effects exerted by triptolide via autophagy imply that autophagy is likely the common pathway through which triptolide exerts its multiple pharmacological activities. 8 -12,15,25 -27
We found that oxidative stress is implicated in the mechanisms underlying triptolide-induced autophagy in HL7702 cells. Oxidative stress is due to the imbalance between oxidants and antioxidants. It results in the accumulation of oxidized components and ultimately disrupts cellular homeostasis. 28 Oxidative stress has been confirmed as a mechanism of the hepatotoxic effects of triptolide. 24 Triptolide exposure drastically increases the generation of ROS, reduces the activities of antioxidant enzymes, and subsequently causes oxidative stress and cell damage in mouse liver. 14 Mitochondria are the major producers of ROS. In rats, triptolide induces mitochondrial membrane depolarization, which results in liver damage accompanied by elevations in ROS contents. 29 Furthermore, triptolide treatment results in the drastic loss of mitochondrial membrane potential and induces mitochondrial swelling in isolated liver mitochondria accompanied by the excessive accumulation of intracellular ROS, which may further damage mitochondria. 2,13 In this study, we found that triptolide could induce oxidative stress through the induction of ROS and the depletion of the antioxidants SOD and GSH, ultimately increasing the level of the lipid peroxidation product MDA in HL7702 cells. However, pretreating HL7702 cells with the ROS scavenger NAC prior to triptolide treatment markedly attenuated the enhanced generation of ROS and partially reversed the upregulation of LC3II. These data indicate that the proautophagic effects of triptolide are dependent on oxidative stress. Indeed, the autophagic pathway is triggered in response to oxidative stress. 30 Similar phenomena have been observed in triptolide-induced myocardial autophagy. Triptolide depolarizes mitochondria, upregulates ROS levels, increases the expressions of Beclin1 and LC3II, and finally induces cardiac injury. Colocalization experiments have proven that dysfunctional mitochondria are selectively transferred into autophagosomal vacuoles for degradation, suggesting that autophagy is cardioprotective through eliminating dysfunctional mitochondria. 12,31 In murine leukemia WEHI-3 cells, triptolide stimulates autophagy by increasing ROS generation on the mitochondrial level. 9
Increased Beclin1 expression likely plays a vital role in the triptolide-induced autophagy of HL7702 cells. Beclin1, the mammalian ortholog of yeast Atg6, forms a complex that has a central role in autophagosome formation. 32 Beclin1 overexpression induces autophagic activity and promotes autophagic flux. 16 Reactive oxygen species–generating agents induce autophagy by regulating Belcin1 expression. 33 Therefore, triptolide likely causes oxidative stress, which is subsequently responsible for autophagic activation by increasing Belcin1 expression. However, further studies are necessary to elucidate the exact role of triptolide-induced oxidative stress in Beclin1 regulation. Several other distinct mechanisms are also involved in ROS-induced autophagy. For example, elevated ROS levels induce autophagy by repressing mTORC1 via the LKB1/AMP-activated kinase metabolic pathway in the cytoplasm. 34 The enhancement of autophagy under oxidative stress is related to the inhibition of cytoplasmic p53. 35 Oxidative stress also upregulates the expression of the autophagy-related proteins p62 and UVRAG, which are mediated by the activation of transcription factors TFEB or NRF2. 36,37 Future work is needed to address whether these machineries or signaling pathways are implicated in triptolide-induced autophagy.
Autophagy plays a crucial role in maintaining cellular homeostasis and is an adaptive response that enables cell survival under stressful conditions. Nevertheless, excessive autophagy exerts adverse effects by promoting cell death under certain circumstances. 38 We used CQ to intervene with autophagy and observed that the activities of caspase 3 and caspase 9 drastically increased. This observation is suggestive of crosstalk between triptolide-mediated apoptosis and autophagy in HL7702 cells. Ample evidence has shown that autophagy engages with apoptosis. For example, autophagy, a bulk degradation process, is responsible for the turnover of damaged mitochondria and regulates hepatic apoptosis via the mitochondrial pathway. 39 Thus, autophagy may serve as a defense mechanism that allows escape from apoptosis and enables hepatic cell survival under triptolide-induced oxidative stress (Figure 6). Several mechanisms participate in the complicated interconnection between autophagy and apoptosis. Altered autophagy proteins may regulate the apoptotic pathway directly, 39 and autophagy and apoptosis share common regulators. 40 Further experiments on the definitive signal pathways that control the triptolide-modulated crosstalk between autophagy and apoptosis in HL7702 cells are warranted.
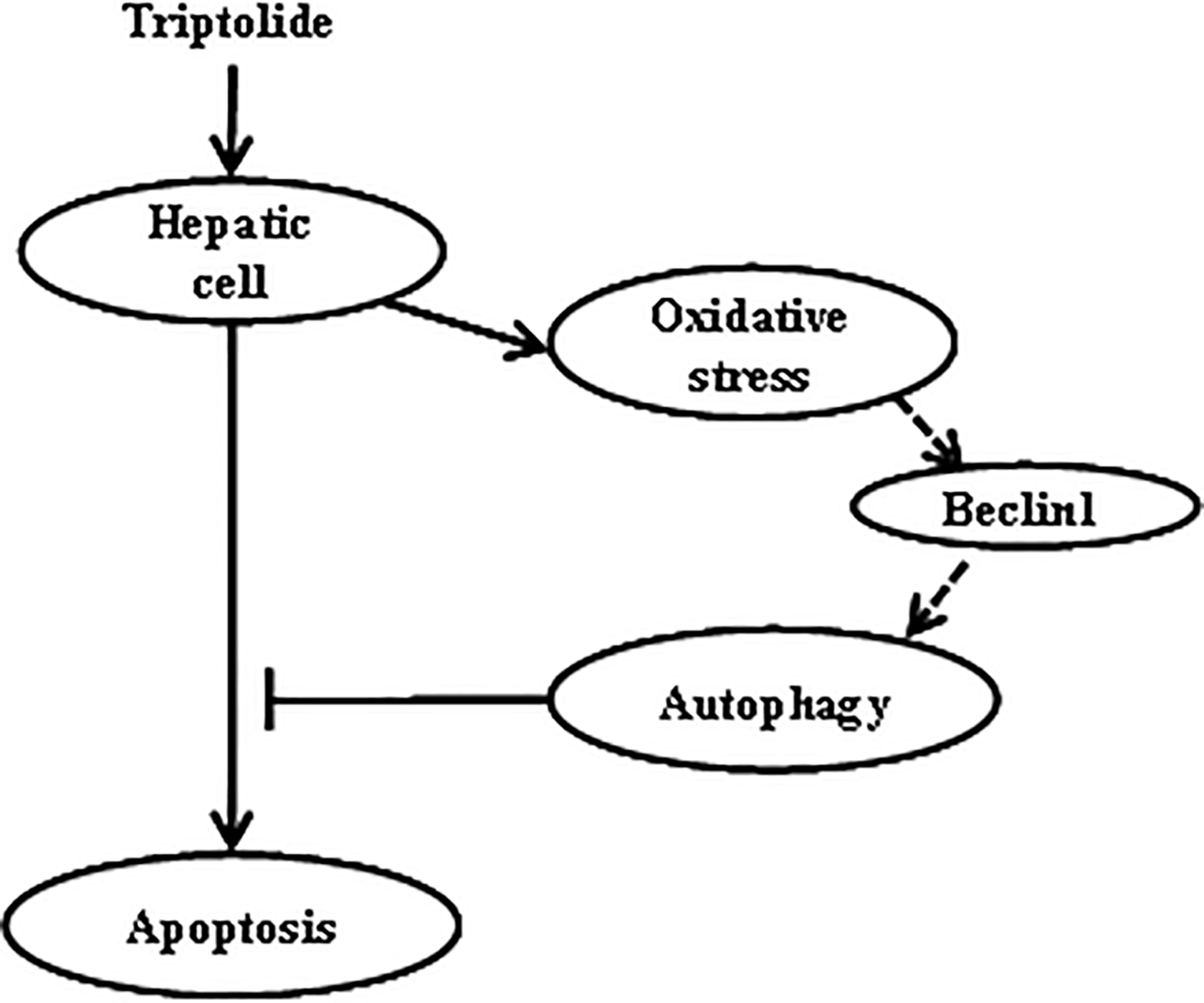
Schematic diagram of autophagy regulating triptolide-induced apoptosis in hepatic cells. Triptolide-induced oxidative stress consequently enhances autophagic activity in hepatic cells, and increased Beclin1 likely participates in it. Autophagy is a protective machinery against triptolide-induced apoptosis as autophagy inhibitor markedly increases the activities of caspase 3 and caspase 9.
In summary, we found that triptolide treatment causes damage in human normal liver HL7702 cells and that autophagy induced through the mediation of oxidative stress functions as a protective mechanism against triptolide-induced cytotoxicity. Our data indicate that the manipulation of autophagy might represent a novel therapeutic avenue for preventing or ameliorating the triptolide-induced hepatotoxicity.
Footnotes
Author Contributions
Y. Wei contributed to conception and design and drafted manuscript. Z. Luan contributed to design and contributed to analysis. B. Liu contributed to design and contributed to analysis. Y. Wang contributed to design and contributed to analysis. Y. Chang contributed to interpretation and critically revised the manuscript. H. Xue contributed to interpretation and critically revised the manuscript. J. Ren contributed to interpretation and critically revised the manuscript. All authors gave final approval and agree to be accountable for all aspects of work ensuring integrity and accuracy.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This study was sponsored by the Applied Basic Research Program of Science and Technology Department of Shanxi Province (No. 201801D121357), the Research Project of Shanxi Provincial Health and Family Planning Commission (No. 201601114), and the Research Fund from Shanxi Key Laboratory of Innovative Drug for Treatment of Serious Diseases Basing on the Chronic Inflammation (No. SXIDL-2018-08).