
Abstract
Burn healing should be regarded as a dynamic process consisting of two main, interrelated phases: (a) the inflammatory phase when neutrophils and monocytes infiltrate the injury site, through localized vasodilation and fluid extravasation, and (b) the proliferative-remodeling phase, which represents a key event in wound healing. In the skin, both canonical autophagy (induced by starvation, oxidative stress, and environmental aggressions) and non-canonical or selective autophagy have evolved to play a discrete, but, essential, “housekeeping” role, for homeostasis, immune tolerance, and survival. Experimental data supporting the pro-survival roles of autophagy, highlighting its Yang, luminous and positive feature of this complex but insufficient explored molecular pathway, have been reported. Autophagic cell death describes an “excessive” degradation of important cellular components that are necessary for normal cell function. This deadly molecular mechanism brings to light the darker, concealed, Yin feature of autophagy. Autophagy seems to perform dual, conflicting roles in the angiogenesis context, revealing once again, its Yin–Yang features. Autophagy with its Yin–Yang features remains the shadow player, able to decide quietly whether the cell survives or dies.
Introduction
The skin is a complex organized first line of defense. 1 The skin micro-environment is strongly influenced by temperature, diet, pH, moisture, sebum level, resident immune cells, infectious exposure, and last but not least, oxidative stress.1–4 However, the skin is gifted with an insufficiently explored arsenal of molecular and cellular weapons and able to counterattack potential external threats.1,2 The efficiency of skin protective function relies mainly on molecular mechanisms controlling and sustaining the continuous removal of dead cells and other debris without alarming the immune system.1–4 In this context, one of the main roles will be played by autophagy. 5
Skin cell populations are both non-immune and immune.2,6 Epidermis cells are represented by keratinocytes, Langerhans cells (LCs), dendritic epidermal γδ T cells, melanocytes, and Merkel cells. 7 The dermis has populations of non-immune cells, like fibroblasts, endothelial cells, and neurons and immune cells, such as B cells, macrophages, T cells, innate lymphoid cells, and NK.1–3 Hypodermis consists of adipocytes, lymphatic and blood vessels, and nerves.1–3 Normally, the epidermis and dermis have circulating immune cells (neutrophils, monocytes, macrophage, αβ T cells, γd T cells, NK cells, B cells, and innate lymphoid cells) and non-immune cells (such as keratinocytes, fibroblasts, and melanocytes).1–3
On the other hand, in the skin, both canonical autophagy (induced by starvation, oxidative stress, and environmental aggressions) and non-canonical or selective autophagy have evolved in order to play a discrete, but, essential, “housekeeping” role, in homeostasis, immune tolerance, and survival.1,6 The skin is exposed to many and various environmental stressing factors, so it has big energy and resource requirements. However, being a nutrient-poor organ, its functions and survival mainly depend on the recycling of limited resources via the complex autophagy system.5,8
Regarding autophagy, of all the organs of the human body, the skin remains one of the less studied. The aim of our review is to shed some light upon the vailed roles of autophagy in burn wounds healing, and, also, to outline its Yin Yang duplicitous features, as a pro-survival/cell death initiator mechanism, in burns context. We have focused on three main topics: (1) the molecular mechanism of the autophagic process, (2) autophagy as a pro-survival mechanism in burns, the Yang, and (3) autophagy as a cell-death promotor pathway in burns, the Yin.
Autophagy
The word “Autophagy” is derived from the ancient Greek language, and it means self (auto) eating (phagy). The term autophagy was first presented by Christian de Duve, who won the Nobel Prize in Medicine for studying lysosomes, in 1974.9–14 Autophagy is a highly conserved molecular pathway across eukaryotes’ evolution. This molecular machine enables the cells to recycle cellular debris via lysosomes, ensuring, in this way, survival during periods of nutrient deprivation and stress. 9 However, presently, it becomes clearer and clearer that the pathway of autophagy is intimately involved, not only in cell adaptation to starvation but also in inflammation, apoptosis, and cellular necrosis. The autophagy pathway can be classified as follows: macroautophagy (canonical autophagy—known as autophagy), microautophagy, and chaperone-mediated autophagy.9,11,15 Characteristic of macroautophagy is the autophagosome formation. The autophagosome represents a double membrane vesicle, able to engulf cytosolic proteins, damaged organelles, and other cellular materials.9,11,15
Microautophagy represents the substrate translocation, via direct protrusion or invagination, into lysosome, for degradation.9,11,15 The chaperone-mediated autophagy involves the direct translocation of the substrate proteins across the lysosomal membrane, by a chaperone protein Hsc70 (heat shock cognate 70)-mediated mechanism.9,11,15
The molecular orchestra of autophagy is precisely controlled by the ATG protein group.9,16 Briefly, the main steps of the autophagic pathway are (1) the pre-initiation complex organization; (2) the phagophore formation; (3) autophagosome elaboration; (4) autophagosome–lysosome fusion triggering the autolysosome formation; and (5) cargo degradation (Figure 1).9,16–18 Main steps of the autophagic pathway.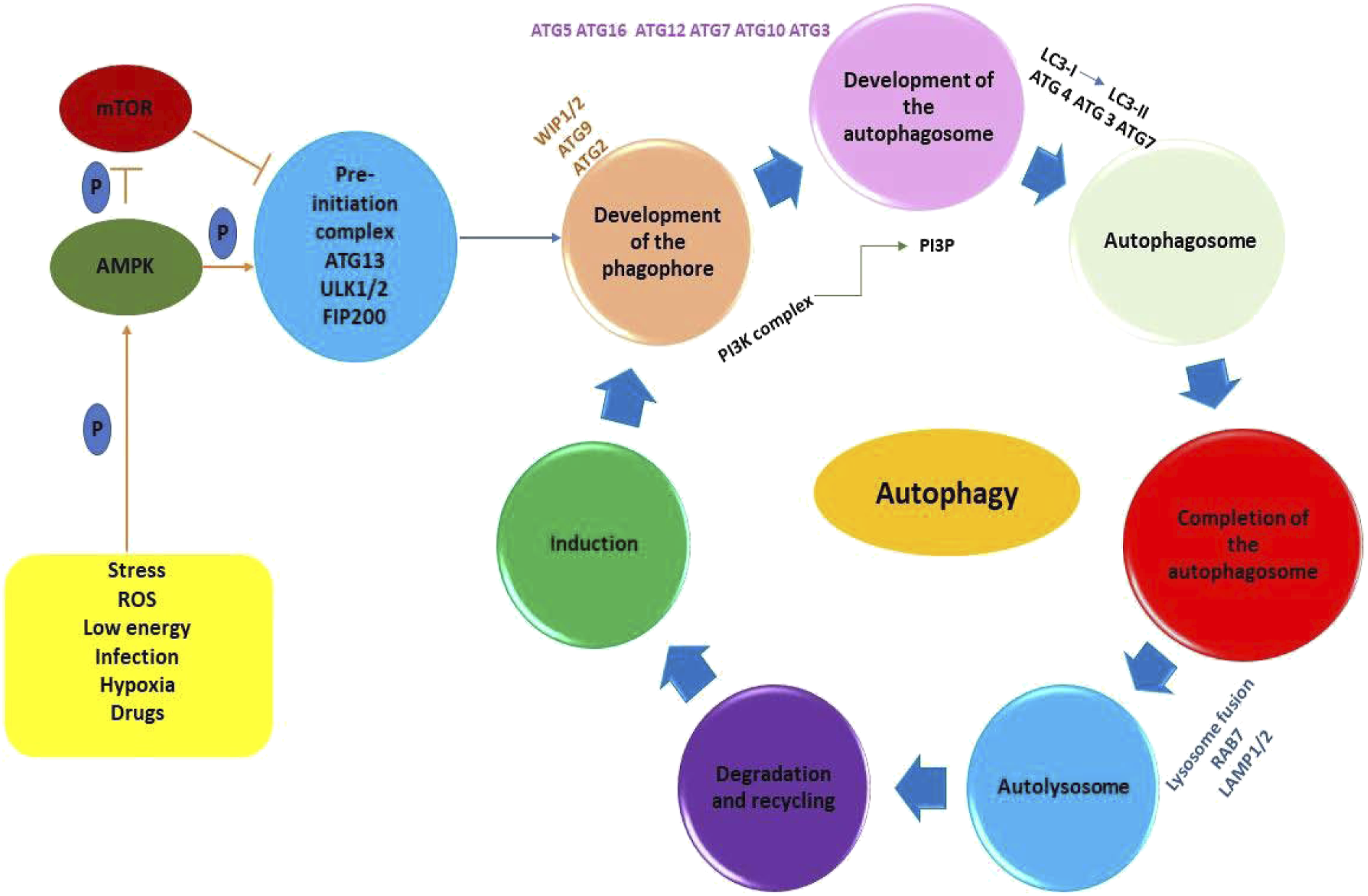
The mammalian target of rapamycin (mTOR) represents the key regulator of autophagy initiation; more precisely, mTOR inhibition triggers autophagy induction by the assembly of ULK1/2, ATG13, and FIP200, to elaborate the pre-initiation complex, in the presence of unwanted cellular debris (mitochondria, pathogens, and protein aggregates, representing the cargo) (Figure 1).15–17 This molecular event will, in turn, activate the Class III phosphatidylinositol-3-kinase (PI3K) complex, formed by ATG14 (UVRAG)-VPS15-VPS34-Beclin1. The main function of this complex is to recruit the ATG proteins to the autophagosome assembly site.15–17 During autophagosome elongation, E3 (Ubiquitin)-ligase ATG7 is recruited to the autophagosome membrane and triggers the ATG5–ATG12–ATG16L1 complex generation.1,16,19 E2-like enzyme ATG3 generates the ATG12–ATG3 conjugate, controlling mitochondrial homeostasis.1,16,19 ATG7 can recruit ATG3 and ATG10 leading to ATG7–ATG3 and ATG10–ATG3 complexes, respectively.17,19,20 ATG12-conjugation is vital for pre-autophagosomes formation.20,21 ATG3 is involved in the LC3-I conjugation with phosphatidylethanolamine (PE). LC3 lipidation with PE forms LC3-II, necessary for the autophagosomes complete assembly.19–21 LC3-PE is included into the mature autophagosome.19–21 The completed autophagosome finally is able to fuse with the lysosome, resulting the autolysosome inside which the cargo is degraded by lysosomal hydrolases. The degradation products are released back into the cytosol and recycled.19–21
As mentioned above, mTOR is a critical conductor of autophagy initiation. Mitogen-activated protein kinase (MAPK) and protein kinase B (Akt) activate mTOR via the interaction of tuberous sclerosis complex (TSC) 1/2, Rheb, and the mammalian target of rapamycin complex 1/2 (mTORC1/2). 10 ATG13 and the serine–threonine kinase ATG1 phosphorylation are inhibited by the activated mTOR, suppressing in this way the initiation of the autophagy pathway. 16
Autophagy represents a complex molecular pathway which should not be considered limited only to cell survival during starvation. In reality, autophagy plays key role in regulating important cellular events.9,22–24 This highlights the importance of investigating autophagy’s hidden influences on various biological mechanisms, in different contexts, such as burn wound healing. The inflammatory response in the skin, induced by environmental irritants, such as burns, involves autophagy as a complex regulator of specific molecular events. 1
Autophagy: a pro-survival mechanism—the Yang
Compared to other types of wounds, burn wounds are a special type of skin lesions, in many ways: molecular events, signaling pathways involved, pathophysiology, and the entire, imposed management. 25 The burn wound healing should be regarded as a complex and dynamic process, involving innate immune cells (neutrophils, monocytes, and macrophages), adaptive immune cells (alpha beta (αβ) T cells and the gamma delta (γd) T cells), and non-immune cells (keratinocytes, fibroblasts, mesenchymal stem cells, and smooth muscle cells).25,26
Autophagy levels during burn wound healing main steps.


The most important steps of the burn wound healing are briefly illustrated in Figure 2. The most important steps of burn wound healing.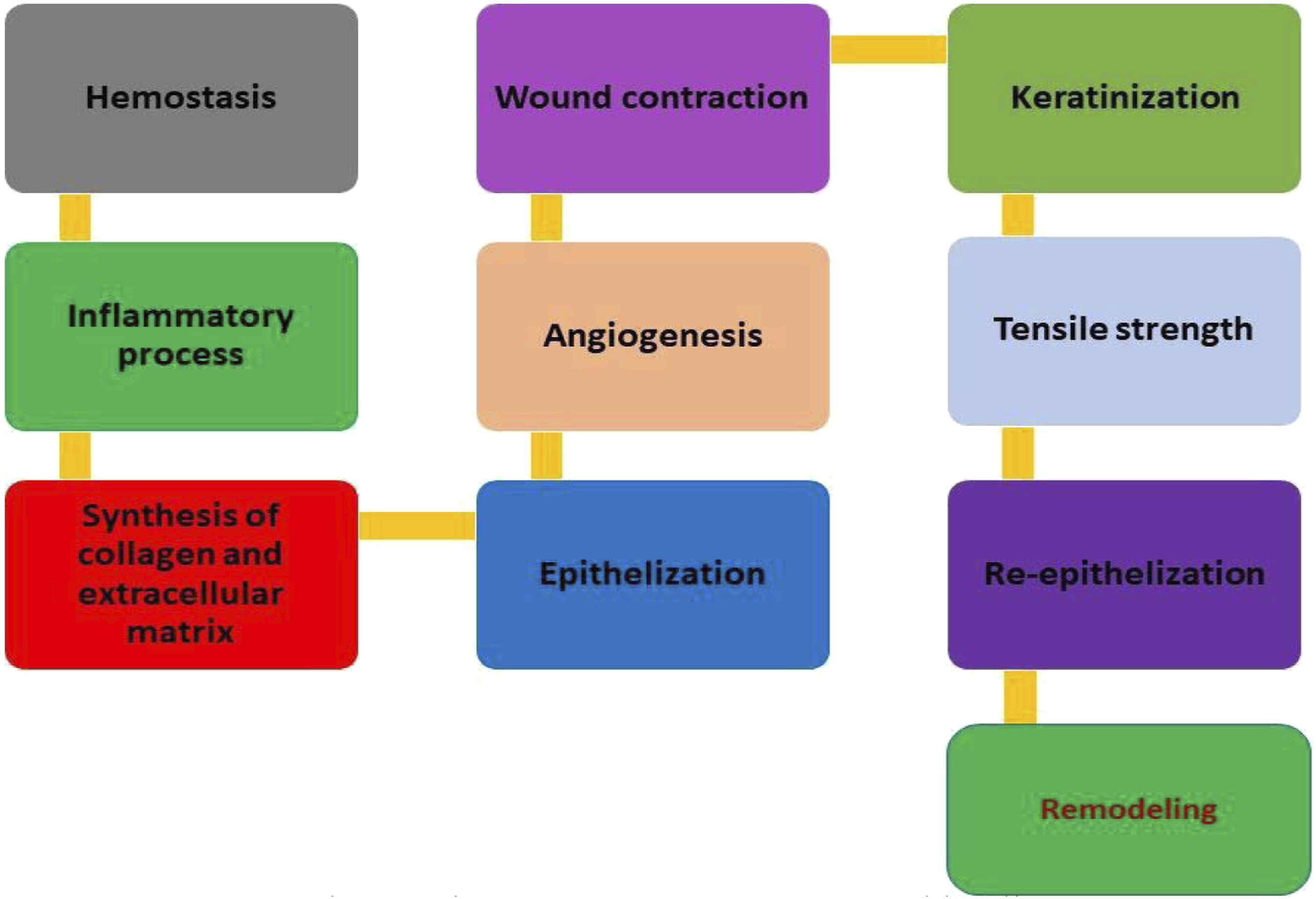
Autophagy and the inflammatory phase of burn wound healing
Immediately after the thermal injury occurred, the burn wound presents three zones: (1) the coagulation zone (most damaged in the central portion); (2) the stasis zone or zone of ischemia; and (3) the hyperemia zone (characterized by increased inflammatory-induced vasodilation).1,27,28
Hemostasis is initiated immediately after the thermal injury occurred and involves platelets recruitment and aggregation; vasoconstriction; secretion of clotting and growth factors (like platelet-derived growth factor (PDGF), epidermal growth factor (EGF) and transforming growth factor-β (TGFβ)) by platelets, macrophages, and fibroblasts, triggering the fibrin clot formation at the thermic injury site. The fibrin clot will serve as a provisional matrix for the next steps of the healing process.25–28
After the thermal injury occurred, neutrophils, monocytes, and monocyte-derived macrophages M1 recruited to the lesion site due to localized vasodilation. These immune cells will practically initiate and amplify the inflammatory phase.25–28 The macrophages remove cell debris and pathogens from the injury site. Neutrophils and macrophages release cytokines (tumor necrosis factor (TNF); IL-1, IL-8) and growth factors (insulin-like growth factor (IGF) and vascular endothelial growth factor (VEGF)).25–28 Autophagy plays discrete, but, however, essential roles in the molecular basis of these immune cells’ activity. Moreover, autophagy should be regarded as a complex target of the molecular signals represented by cytokines (Figure 3).1,25–29 The complex interrelations between cytokines and autophagy.
As illustrated in Figure 3, autophagy is induced by interferon (IFN)-γ, interleukin IL-1β, IL-α, and tumor necrosis factor (TNF)-α.28,29 Important to emphasize is that the autophagy relationship with IL-α, IL-1β, IL-17, and TNF-α can be influenced by different conditions, such as a burn wound.28,29
Bhaskaran outlined that IL-1b could modulate Akt/mTOR metabolic signaling proteins in T cells. Moreover, these authors have shown that IL-1b increased p-Akt and p-mTOR expression in naïve and Foxp3+CD4+T cells. 30
The PI3K/AKT/mTOR signaling pathway represents a key modulator of autophagy. More precisely, the PI3K/AKT/mTOR pathway activation suppresses autophagy and promotes inflammatory responses in several diseases. 31 Tang et al. have shown that TNF-α-induced PI3K/AKT/mTOR signaling activation has been abolished by lncRNA MEG3 (long non-coding RNA maternally expressed gene 3) overexpression in keratinocytes. Moreover, PI3K/AKT/mTOR pathway inhibition triggered the downregulation of TNF-α-induced inflammation and restored the autophagy level. 31
The autophagic machine is blocked by IL-4, IL-10, IL-13, and IL-33.
In turn, autophagy induces the synthesis and secretion of IFN-γ, TNF-α, and IL-1β and abolishes the TNF-α, IL-17, IL-1β, and IL-α release.28,29
Neutrophils are the best represented granulocytes and are considered real attack pawns of the immune system. 32 Autophagy role in the neutrophils function is highlighted by the reported data revealing that autophagy-deficient neutrophils showed NADPH-oxidase-mediated reactive oxygen species (ROS) production, impaired degranulation, and abnormal inflammatory responses. 30 Moreover, the neutrophils from leprosy patients’ skin showed intensified autophagy and exhibited accelerated apoptosis in vitro. 33 In neutrophil-mediated inflammation, autophagy represents a protective mechanism.
Neutrophil autophagy blocking may trigger an uncontrolled inflammatory response. 34 It has been reported that autophagy decreases cytokine production and downregulates neutrophil influx.35,36 Autophagy decreases degranulation and ROS generation, triggering apoptosis downregulation.37,38
Macrophages are phagocyting cells, located in the epidermis and dermis. 6 In normal conditions, these cells are involved in maintaining the skin immunotolerant environment.6,39
In macrophages, autophagy also plays key roles regarding the functions of these phagocytic cells. These important roles are outlined by the experimental data revealing that in leprosy patients, skin macrophages presented significant upregulations of autophagy genes, especially atg14 and beclin1. 40
Other important immune cells, deeply involved in the burn wound evolution, are the adaptive immune cells, the alpha beta (αβ) T cells and the gamma delta (γd) T cells. The alpha beta (αβ) T cells (CD8+ T cells and CD4+ T cells) are maintained in the skin long after the immune response is over.41–48 Atg7-deficient (αβ) T cells are not able to participate in the skin homeostasis maintenance,49,50 illustrating once again the importance of the autophagic pathway in sustaining their functions. At the same time, gamma delta (γd) T cells are found in the epidermis and dermis and are named dendritic epidermal γd T cells (DETCs) and dermal γd T cells, respectively.51–53 Like αβ T cells, they have mediator roles in tissue repair.51,54 γd T cells are among the first responders to skin damage, including burns. This type of T cells is important in maintaining skin homeostasis in different conditions, including burns, by releasing cytokines like IGF-1, TNF-α, and KGF-1. These cytokines underlie their roles in burn wound repair. 54 DETCs develop close contacts with keratinocytes and have a significant contribution in wound healing.55,56 Interestingly, skin γd T cells are able to sustain autophagy for survival, also in the absence of cytokines.54,57 These mentioned experimental data highlight the important roles performed by autophagy in the molecular landscape of the functions of immune cells like neutrophils and macrophages, especially considering that they are the key players in the inflammatory phase initiation of burn wound healing.
Autophagy, a complex and dynamic molecular flux, represents a key point for cell survival.58,59 As mentioned above, cytosolic LC3 (LC3-I) conversion to the autophagosome-associated form LC3-II by conjugation of phosphatidylethanolamine is considered an essential event for autophagy initiation.60,61 P62 should be regarded as another autophagy biomarker since after the autophagic machinery starts, p62 is degraded.58,59 Beclin-1 level can also be used as an indicator of autophagy.60,61 Putting all this together, LC3-II and Beclin-1 increment along with p62 decrease should be regarded as biomarkers of autophagic activity level during burn wound evolution. Heba A et al. reported experimental data supporting the pro-survival role of autophagy, highlighting its Yang, luminous, and positive feature of this complex but insufficient explored molecular pathway. 61 The study of Heba, conducted on skin samples from burn wounds, revealed, by measuring LC3-II and Beclin-1, a significant decrease of autophagic level (p < .001) during the first 24 h. 61 After 24 h, autophagy intensity began to increase, but it did not reach the normal level up to 72 h after the burn injury occurred. 61 This increase took place in accordance with the epithelial cells’ migration and scab formation. 61 These findings outlined the hypothesis that, in the very early stage of the burn injury, the cell number sustaining autophagy is reduced, necrosis becoming the main event. 61 After 24–72 h, when the tissue necrosis decreased in intensity, but the surrounding tissue presented ischemic damages as a consequence of strong inflammatory signals, autophagy reenters the scene, as a molecular pro-survival mechanism, being able to protect cells against these stress effects. This hypothesis may explain the later increase of autophagy intensity, illustrated by the LC3-II and Beclin-1 increase. 61
It has been shown that excessive exposure to UV radiation results in acute skin damage including epidermal injuries. These injuries trigger an influx of activated immune cells into the wounded skin bed generating a localized inflammatory response that further exacerbates inflammation, altering the tissue repair process.62,63 Das et al. have shown that a single dose of vitamin D decreased the UV-induced skin inflammation and was sufficient to sustain skin cell survival and to accelerate the tissue recovery process. 64 Strozyc et al. have highlighted that the uncontrolled cell death caused by excessive UV exposure may be offset by survival signals transmitted from upregulated autophagy. 65 Autophagy is regarded as a powerful immune regulator able to counteract infection and respond to toll-like receptor signaling, directing the cells towards survival or apoptosis.66–68 Moreover, it has been reported that macrophages autophagy upregulation had protective effects from acute and chronic organ injury through reducing inflammation intensity, promoting cell survival, and, finally, supporting the tissue repair process.69–72 Jiang et al. have also shown that enhanced autophagy had protective effects against polymicrobial sepsis by dampening the cytokine storm triggered by microbial load. 73
These studies support the conclusion that the enhanced autophagy represents a very important pro-survival mechanism, influenced, however, by the surrounding micro-environment and stress response. However, future studies are needed in order to clarify the role of vitamin D, via autophagy, in burn wounds’ recovery.
Autophagy and the molecular landscape of the proliferative and remodeling phase
The next phase of burn wound healing is the proliferative and remodeling phase.28,61 The molecular events characterizing this phase are strongly based on fibroblasts, keratinocytes, and endothelial cells recruitment, activation and proliferation at the wound site.28,61 The proliferation of these non-immune cells insures the provisional matrix substitution with a connective tissue matrix (Table 1).28,61 The important next steps are represented by angiogenesis, granulation tissue formation, and epithelialization (Table 1).28,61
Keratinocytes are non-immune cells involved in both angiogenesis (restoring the blood vessels and resuming circulation) and epithelialization (which means wound surface closure) modulation of angiogenesis through circadian oscillation of vascular endothelial growth factor A (VEGF-A) in epidermal keratinocytes.1,74,75
Keratinocytes represent the foundation of the epidermis. From all the skin cells, they are the most studied. Human keratinocytes are able to initiate inflammasomes assembly upon either UVB irradiation, viral infection, or, probably, burns.1,74 The keratinocyte growth factor (FGF7/KGF) controls keratinocyte differentiation and autophagy initiation. Keratinocyte FGF7/KGF-controlled differentiation triggers the LC3 expression and autophagy initiation via the PI3K–AKT–mTOR pathway,76,77 lysosomal enzyme activation, and cellular components degradation.1,6,78–80 In the case of Atg7-deficient keratinocytes, N,N′-dimethyl-4,4′-bipyridinium dichloride (paraquat) treatment led to p53 and p21 accumulation and abnormal cellular aging, 79 highlighting the importance of the autophagic machinery. Reported experimental data revealed that Atg5-deficient keratinocytes are unable to undergo differentiation.1,81–83 Regarding keratinocytes, autophagy reveals again its bright, Yang feature.
The resident fibroblasts are transformed into myofibroblasts, which will contribute to extracellular matrix (ECM) deposition. 1 The final steps of the proliferative and remodeling phase are granulation tissue becomes mature and the ECM is remodeled by matrix metalloproteinases (MMPs) under the precise control of growth factors and tissue inhibitors of metalloproteinases (TIMPs), leading to increased tensile strength (Figure 2). 1
Angiogenesis involves endothelial cell activation by growth factors like FGFs, VEGF, and hepatocyte growth factor (HGF). 84 Angiogenesis, a key pathway in wound healing, presents the following steps: (1) the surrounding basement membrane degradation by endothelial proteolytic enzymes; (2) the initiation of the sprout formation; (3) endothelial cell proliferation and migration; and (4) tube-like structure formation by the migrating cells.85,86
All these are sustained by Liang et al. findings, highlighting that angiogenesis was stimulated during the heat-denatured endothelial cell (HDEC) recovery. 84 Angiogenesis initiation has been illustrated by increased endothelial cell proliferation, migration, and tube organization. 87 Liang et al. also pointed, based on their experimental results, that (1) autophagy level increments during HDECs recovery depended on intracellular ROS (reactive oxygen species) generation; (2) autophagy inhibition suppressed endothelial cell proliferation, migration, and tube-like structure formation, in vitro; (3) autophagy proved to be vital for pro-angiogenesis during HDECs recovery, in vivo; and (4) intracellular ROS are subtle but essential regulators of AMPK/Akt/mTOR signaling, enhancing the autophagy level and initiating angiogenesis during HDEC recovery. 84
The post-burn inflammatory phase is characterized by a huge ROS generation, triggering the progression of the local and, also, distant inflammatory reactions. 88 Immediately after the thermal injury, PMNs invade the lesion scene inducing the release of massive amounts of ROS in the interstitial fluid. However, the antioxidant enzyme concentration and activity in the wound fluid is modest and insufficient in order to remove the large ROS amounts, generated during the post-injury phase.
Reactive nitrogen species (RNS) and ROS are highly reactive species released during the cellular metabolism, in both normal and pathological conditions. Basal ROS/RNS levels play essential homeostatic roles in regulating the molecular signaling pathways, involved in metabolism control, proliferation, and survival.38,89 However, when the redox balance is dysregulated and antioxidant defense systems are surpassed, oxidative stress is initiated. When oxidative stress defeats the cell capacity to repair oxidatively damaged biomolecules (nucleic acids, lipids, and proteins), oxidative damage is initiated. It has been highlighted that oxidative stress triggers autophagosomes accumulation in different types of somatic cells.18,90 However, the precise redox events involved remain unclear. It has been reported that ROS are associated with autophagy induction in starvation conditions.91–93 Oxidative stress-activated autophagy is crucial in protecting cells from apoptosis.94,95 Autophagy impairment will induce and/or increase the oxidative stress. 96 Furthermore, antioxidant molecules are able to suppress autophagy initiation, moderately or completely. 97 In conclusion, ROS not only induce the autophagic pathway but also inhibit it, ROS and autophagy being mutually influenced. ROS, known as key signaling molecules, are very important players in the molecular landscape of angiogenesis, controlling indirectly the endothelial cells’ proliferation and migration. 98
Recent data highlighted the significant roles of ROS in the complex control mechanism of autophagy. 99 Moreover, experimental data revealed the autophagy protective effects against oxidative stress–induced cell death. For instance, it has been shown that the vascular smooth muscle cell platelet-derived growth factor has protective effects against oxidative damage of molecules and 4-hydroxynonenal induced cell death, by upregulating autophagy. 100 In endothelial cells, autophagy induced by glycolysis inhibition with 2-deoxy-D-glucose is controlled by AMPK activation through ROS formation. 101 Reoxygenation-induced ROS generation also triggers autophagy upregulation. The same study revealed that autophagy inhibition increases apoptotic cell death of primary hepatocytes. 102
Liang et al. revealed that both heat treatment and recovery significantly stimulated intracellular ROS generation. 103 They also have shown that ROS generation inhibition by N-acetylcysteine (NAC) triggered the reduction of autophagy in HDECs. 100 More precisely, NAC treatment significantly inhibited AMPK phosphorylation and stimulated AKT and mTOR phosphorylation. This way, autophagy and, consequently, angiogenesis were inhibited during the recovery of HDECs in vivo. 104 These data lead to the conclusion that in the HDECs recovery context, autophagy and angiogenesis are interconnected by a fine molecular network of ROS. ROS are able to initiate autophagy through various signaling pathways, including AMPK and mTOR pathways, playing crucial roles.84,105
In response to metabolic stress, AMPK is one of the main actors on the autophagy regulation scene in endothelial cells.104,105 AMPK, as a positive autophagy regulator, downregulates the AKT/mTOR pathway,106,107 one of the main modulators of autophagy. AKT regulates autophagy mostly via mTOR activity modulation. AKT pathway initiation by the recombinant active human AKT1 full-length protein (rAKT) was able to inhibit autophagy, affecting the angiogenesis process, normally induced in HDECs by autophagy.84,103 However, future research will have to clarify the molecular mechanisms through which ROS induce and support the pro-survival feature of autophagy during burn wound evolution and healing.
Autophagic cell death—the Yin
Usually, autophagy initiation in response to stress represents a pro-survival molecular mechanism. However, in some specific situations, autophagy changes its protective role and becomes the mediator and inducer of the autophagic cell death. 108 We still know very little about the autophagy roles in the evolution of burn wounds. Recent studies revealed that in case of burns, cell death may occur due to necrosis, autophagy, and apoptosis, all leading through a specific molecular path to burn injury positive or negative evolution.109–112
Necroptosis, necrosis, and secondary necrosis following apoptosis have been recently highlighted as different mechanisms of cell death.109–112 These mechanisms are based on similar cellular and molecular events: redox imbalance, oxidative burst, hyperpolarization of the mitochondrial membrane, and permeation of lysosomal membrane, and of the cell membrane.109–112
Necrosis is considered an accidental type of cell death, occurring as a response to severe cell damages, like those occurred in the first moments of thermic burns.109–112 The necrotic cell death molecular mechanism can be finely orchestrated by specific signal transduction pathways, involving both the receptor interaction protein kinase 1 and 3 (RIP1 and RIP3). 113 Catabolic processes (necroptosis) also play important roles on cellular necrosis stage. Necrostatins are able to specifically inhibit cellular necrosis. 110 Secondary necrosis represents a form of cellular necrosis that usually occurs in apoptotic cells that escape phagocytosis.112,114 Autophagic cell death describes an “excessive” degradation of important cellular components that are necessary for the normal cell function.115–117 This deadly molecular mechanism brings to light the darker, concealed, Yin feature of autophagy.
ROS, like hydrogen peroxide, superoxide radical, and hydroxyl radicals, are known as key mediators of progressive tissue damage after initial burn injury. 118 The high ROS levels in the burn wound might be caused directly by the thermal energy of burns 119 and, also, by xanthine oxidase and NADPH oxidase enhanced activities.120,121 It has been highlighted that in the zone of stasis, ROS may be involved in the cell death molecular mechanism, possibly via an excessive upregulation of the autophagy pathway.122–125 A possible molecular process used by the autophagic cell death machinery is lysosomal membrane permeation as an answer to stressing factors, like thermic injuries.122–125 Recent studies outlined the hypothesis that ROS are pawns with decisive roles in the fate of the molecular match of autophagic cell death.114–116 It has been shown that the released lysosomal cathepsins have been involved in the oxidative stress–induced apoptosis.122–125 Moreover, since lysosomes represent important sources of ROS, they might play important performances in the redox imbalance initiation and the exacerbation of oxidative stress, triggering oxidative cellular damages.126–129 However, the complex relationships between the disrupted redox balance, oxidative stress and autophagy, and their consequences on burn wound evolution still must be clarified. Many recent research studies focused on investigating the efficacy of different antioxidant agents in burn wound healing progression. Deniz et al. have shown that NAC treatment 1 hour after burns prevented an unfavorable progression of burn wound. NAC is a precursor to reduced glutathione, which has previously been shown to prevent necrosis in the zone of stasis. 130 Starting from these studies, it could be speculated that the antioxidant treatment would be useful only as long as autophagy reveals its Yin feature as a cell-death promoter mechanism. If the autophagic machinery functions as a pro-survival mechanism, the antioxidant treatment may not represent an advantage, if antioxidant species, by reducing ROS levels, could, moderately or completely, trigger autophagy repression. 131 Accumulation of damaged proteins may be responsible for the harmful effects of autophagy suppression.132–135
Tan et al. experimental results revealed higher autophagy rates compared to apoptosis in hair follicle epithelium during the first 24 h after burn injury occurred. 60 They have concluded that in the zone of stasis, both pathways led to cell death but with a different timing, suggesting that two different treatment strategies should be used in order to target both processes. Xiao et al., contrary to the results of Tan et al., reported decreased autophagy rates early in burn injury progression and higher autophagic levels later. 136 The authors have also outlined that rapamycin increased the autophagic rate and, consequently, improved wound healing. These experimental findings suggested that autophagy should be regarded as a key player in preventing burn wound unfavorable progression. 95 In the burn wound context, autophagy proved to be both destructive and protective for the cell, possibly depending on the timing from initial injury, the degree of cell damage, and the evolution of cellular ROS levels.
Future research to discover the autophagy roles in the zone of stasis will be extremely important in order to establish whether the therapeutic strategies should be based on enhancing or inhibiting this complex molecular pathway.137,138
Examples of Yang and Yin, respectively, features of autophagy.

In summary, ROS-induced autophagy in burns could play either a protective role, relieving oxidative stress, or a destructive one. Deciphering the complex ways by which autophagy is controlled via the variations of the cellular redox potential should be considered crucial for future therapeutic strategies development in burns.
The complex dual role of autophagy is still a matter of debate. The predominant manifestation of one of the two contradictory roles (the one that promotes cell survival or the one that induces apoptosis) may depend on the conditions generated by a specific cellular context. It has been clearly highlighted that strictly controlled and balanced lower levels of redox signaling are crucial for normal autophagy. 143 The big question mark arises when the redox status becomes imbalanced and, consequently, disrupts the signaling network necessary to control and modulate autophagy.
In the context of burn wound healing, more and more evidence sustains the potential communication channels between the enigmatic autophagic machinery and skin immune and non-immune cells, against the background of a local inflammatory state that will resonate in the whole body. However, the molecular mechanisms that could clarify the reason for dual features of autophagy in burns and the roles of these communication channels are not yet understood.
According to our knowledge, exploring the specialized literature, we have noticed that until now, there are very few publications that have focused on the study of autophagy in the cells from thermal burn injuries. This reduced number of studies represented one of the starting points in the elaboration of our article. The purpose of our article is to outline more visibly the possible importance of autophagy in the context of thermal burn wounds, especially due to its dual role, demonstrated in other pathological situations, on which the evolution of the healing process could depend. Also, this small number of studies must be considered one of the limitations of our review.
Conclusions
We hope that all these findings presented here had a little contribution to a better understanding of this mysterious and duplicitous molecular machinery, autophagy, in the context of burn wound healing. There are still some mysteries regarding the special molecular mechanisms by which oxidative stress and autophagy mediate and control cell survival in very stressful conditions, such as burns. Understanding these complex mechanisms will help clinicians to establish new starting points for designing accurate therapeutic approaches in burns. Till then, autophagy with its Yin–Yang features remains the shadow player, able to decide quietly whether the cell survives or dies.
Once Edgar Allan Poe said “The boundaries which divide Life from Death are at best shadowy and vague. Who shall say where the one ends, and the other begins?”. Returning to the molecular landscapes, the answer is much less poetical and may be: it depends on the delicate equilibrium between the Yin and Yang features of AUTOPHAGY.
Footnotes
Author Contributions
All authors have equal contributions to the article.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.