
Abstract
Methamphetamine (METH), a highly addictive drug used worldwide, induces oxidative stress in various animal organs, especially the brain. This study evaluated oxidative damage caused by METH to tissues in CD-1 mice and identified a therapeutic drug that could protect against METH-induced toxicity. Male CD-1 mice were pretreated with a novel thiol antioxidant, N-acetylcysteine amide (NACA, 250 mg/kg body weight) or saline. Following this, METH (10 mg/kg body weight) or saline intraperitoneal injections were administered every 2 h over an 8-h period. Animals were killed 24 h after the last exposure. NACA-treated animals exposed to METH experienced significantly lower oxidative stress in their kidneys, livers, and brains than the untreated group, as indicated by their levels of glutathione, malondialdehyde, and protein carbonyl and their catalase and glutathione peroxidase activity. This suggests that METH induces oxidative stress in various organs and that a combination of NACA as a neuro- or tissue-protective agent, in conjunction with current treatment, might effectively treat METH abusers.
Introduction
Methamphetamine (METH) is a potent addictive psychostimulant being abused by approximately 35 million people worldwide. 1,2 It has been reported that approximately 5.8% of Americans, aged 12 years or older, have used METH at least once in their lifetime. 3 A dramatic increase in METH-related emergency department visits is alarming, with >50% involving young adults aged 18–34 years. 4 The relative ease of METH’s availability, coupled with its toxicity, has resulted in increased numbers of associated medical complications and fatalities. 4,5 Chronic use and acute METH intoxication can cause substantial medical consequences, including kidney (rhabdomyolosis, myoglobinuria, and acute renal failure 6 –13 ), liver (acute hepatic failure 14 –17 and centrilobular liver damage 18 ), lungs (pulmonary edema and shortness of breath 19 ), cardiovascular (tachycardia, atrioventricular arrhythmias, myocardial ischemia, and hypertension 19 –21 ), cerebrovascular (hemorrhages, strokes, and seizures 22 –24 ), and psychiatric problems (psychosis, persecutory delusions, persistent visual and auditory hallucinations, depression, anxiety, aggressiveness, social isolation, psychomotor dysfunction 23,25,26 , and suicidal ideation 27 –29 ).
The adverse effects of METH on the central nervous system (CNS) are well recognized and neuropathological changes in the brains of METH abusers are associated with neuropsychiatric problems. 26 METH toxicity occurs via dopamine-dependent reactive oxygen species (ROS) production and oxidative stress. 30 The generation of ROS and nitric oxide has been implicated in METH-induced neurotoxicity. 31 –35 When ROS production overwhelms the antioxidant defense mechanism, it leads to a condition known as oxidative stress. Involvement of ROS in METH toxicity is supported by several studies which reported decreased glutathione (GSH) levels, reduced levels and activities of antioxidant enzymes, and increased lipid peroxidation and protein carbonylation, all hallmarks of oxidative stress. 36 –46
Even though the adverse effects of METH on the brain are well known and are linked to oxidative stress, little information is available about the oxidative stress induced by METH in other organs. Some studies have indicated that METH exposure induces oxidative stress in a rat’s retina and adversely affects the dopaminergic system of the retina as well, 47 particularly during CNS development. Tokunaga et al. 48 studied changes in renal function and found that repeated METH administration induced oxidative DNA injury to the kidney, as a chronic or subacute influence.
Under physiological conditions, tissues are protected against oxidative stress by antioxidant enzymes and small antioxidant molecules, particularly GSH. GSH, a well-known intracellular thiol antioxidant, has been shown to protect cells from various forms of oxidative injury. 49 However, since GSH cannot be transported directly into the cells, the need arises for other compounds that can easily pass into cells and increase intracellular GSH levels. One such compound, known to increase intracellular GSH levels in cells, is the low-molecular-weight thiol antioxidant, N-acetylcysteine amide (NACA). NACA has been shown to be more effective than its parent compound, N-acetylcysteine (NAC), because the neutral carboxyl group of NACA increases its ability to permeate cell membranes and the blood–brain barrier (BBB). This allows NACA to be administered at a lower dose than NAC and prevents many side effects that are generally associated with NAC toxicity. 50 –54
New treatment challenges have arisen with the increased use of METH. 55 However, there are currently no pharmacological treatments for the wide range of symptoms associated with METH-related problems, possibly because of the lack of understanding of METH-induced toxicity. We previously reported that METH causes oxidative stress to BBB cells and also alters the integrity of the BBB by increasing the permeability of the cells. 36 These toxic effects of METH were reversed, however, by pretreatment of the cells with NACA. This antioxidant restored the levels of GSH and scavenged the ROS produced by treatment with METH, thereby maintaining the permeability of the BBB. Considering the ability of NACA to protect BBB cells from METH-induced oxidative stress, the effectiveness of this antioxidant was evaluated as a treatment option for multi-organ oxidative damage caused by METH abuse.
The purpose of the present study was to discover an adjunctive therapeutic drug to prevent or protect against oxidative damage to various organs in METH abusers. To accomplish this, we measured several oxidative stress parameters to determine whether METH induces oxidative stress in organs other than the brain and whether this damage could be prevented by the use of a novel antioxidant, NACA. Observations overall indicated that METH induced oxidative cell damage by increasing lipid peroxidation and decreasing levels of intracellular GSH and the antioxidant enzymes. The use of NACA prevented oxidative damage in the livers, kidneys, and brains of METH-treated animals.
Materials and methods
Materials
CD-1 mice were purchased from Charles River Laboratories International, Inc. (Wilmington, Massachusetts). NACA was purchased from Dr. Glenn Goldstein of David Pharmaceuticals in New York, New York. High-performance liquid chromatography (HPLC)-grade acetonitrile, glacial acetic acid, and o-phosphoric acid were obtained from Fisher Scientific (Pittsburgh, Pennsylvania), while BioRad (Hercules, California) furnished the Bradford reagent. The National Institute on Drug Abuse (NIDA) provided METH, while all other chemicals were obtained from Sigma-Aldrich (St Louis, Missouri), unless otherwise stated.
Animal study design
Male CD-1 mice (7 weeks old) were purchased from Charles River Laboratories International, Inc. (Wilmington, Massachusetts). Animals were housed in a controlled temperature (∼22°C) and humidity (∼55%) animal facility, with a 12-h light and dark cycle. The animals had unlimited access to rodent chow and water and were utilized after 1 day of acclimatization. All animal procedures were conducted under an animal protocol approved by the Institutional Animal Care and Use Committee of the Missouri University of Science and Technology. The mice were divided into four groups: (1) control, (2) METH only, (3) NACA only, and (4) METH + NACA, so that each group contained four mice. All animals were injected (intraperitoneal (i.p)) with either saline, for the control and METH groups or NACA (250 mg/kg body weight) for the NACA and NACA + METH groups, 30 min before exposure to METH (10 mg/kg body weight). This was followed by four more injections of METH every 2 h over an 8-h period. This is one of the most commonly administered doses for acute high-dose METH administration in rodents. 56 This administration protocol provides an adequate model to study overdose pathologies. The dose of NACA (250 mg/kg body weight) was found to be optimum based on our previous studies with NACA, where a range of NACA concentrations was used (250–500 mg/kg body weight). All rats were anesthetized 24 h after the last i.p. METH injection with a 40% urethane solution (0.1 ml/10 g body weight). All mice were weighed at the beginning and the end of the study. After sacrificing the animals, their organs were harvested, rinsed with phosphate buffered saline solution, and then immediately placed on dry ice. Samples were stored at a temperature of −80°C for further analysis.
Experimental design for oxidative stress parameters
Determination of GSH levels
The levels of GSH in the tissues were determined by reversed phase-HPLC, according to the method developed in our laboratory. 57 The HPLC system (Thermo Electron Corporation, Florida) consisted of a Finnigan Spectra System vacuum membrane degasser (model SCM1000), a gradient pump (model P2000), autosampler (model AS3000), and a fluorescence detector (model FL3000), with λ ex = 330 nm and λ em = 376 nm. The HPLC column used was a Reliasil ODS-1 C18 column (5-μm packing material) with 250 mm × 4.6 mm inner diameter (i.d.; Column Engineering, Ontario, California). The mobile phase (70% acetonitrile and 30% water) was adjusted to a pH of 2 with acetic acid and o-phosphoric acid. The N-(1-pyrenyl)-maleimide (NPM) derivatives of GSH were eluted from the column isocratically at a flow rate of 1 ml/min. The tissue samples were homogenized in a serine borate buffer (100 mM Tris HCl, 10 mM borate, 5 mM serine, and 1 mM diethylenetriaminepentaacetic acid), centrifuged, and 50 μl of the supernatant were added to 230 μl of HPLC grade water and 750 μl of NPM (1 mM in acetonitrile). The resulting solution was incubated at room temperature for 5 min and the reaction was stopped by adding 10 μl of 2 N HCl. The samples were then filtered through a 0.45-μm filter and injected into the HPLC system.
Determination of malondialdehyde
The malondialdehyde (MDA) levels were determined according to the method described by Draper et al. 58 Briefly, 550 μl of 5% tricholoroacetic acid (TCA) and 100 μl of 500 ppm butylated hydroxytoluene in methanol were added to 350 μl of the tissue homogenates and boiled for 30 min in a water bath. After cooling on ice, the mixtures were centrifuged, and the supernatant collected was mixed 1:1 with saturated thiobarbituric acid (TBA). The mixture was again heated in a water bath for 30 min, followed by cooling on ice. Five hundred microliters of the mixture was extracted with 1 ml of n-butanol and centrifuged to facilitate the separation of phases. The resulting organic layers were first filtered through 0.45 μm filters and then injected into the HPLC system (Shimadzu, US), which consisted of a pump (model LC-6A), a Rheodyne injection valve, and a fluorescence detector (model RF 535). The column was a 100 mm × 4.6 mm i.d. C18 column (3-μm packing material, Astec, Bellefonte, Pennsylvania). The mobile phase used contained 69.4% sodium phosphate buffer, 30% acetonitrile, and 0.6% tetrahydrofuran. The fluorescent product was monitored at λ ex = 515 nm and λ em = 550 nm. Malondialdehyde bis (dimethyl acetal), which gives MDA on acid treatment, was used as a standard. This method differs from commonly used Thio Barbituric Acid Reactive Substances (TBARS) assay in that it specifically determines TBA-MDA adducts by HPLC.
Catalase activity assay
Catalase (CAT) activity was measured according to the method described by Aebi. 59 Briefly, it was measured spectrophotometrically at a wavelength of 240 nm, in the supernatant of the tissue homogenate, following the exponential disappearance of hydrogen peroxide (H2O2, 10 mM). The CAT activity was calculated from the equation A 60 = A initial e −kt , where k represents the rate constant, A initial is the initial absorbance, and A 60 is the absorbance after 60 s have passed.
Glutathione peroxidase activity
Glutathione peroxidase (GPx) activity was determined using a glutathione peroxidase colorimetric assay kit purchased from Oxis Research (Foster City, California). The tissue samples were homogenized in 50 mM of phosphate buffer (pH 7.4), containing 1 mM ethylenediaminetetraacetic acid, which were then centrifuged at 7500g for 10 min. The resulting supernatant was collected to be used for the assay, while the remaining debris was discarded. In brief, the assay buffer, supernatant, and NADPH reagent (containing glutathione reductase, GSH, and NADPH) were placed in a cuvette and the reaction was initiated by the addition of t-butylhydroperoxide. The decrease in absorbance at 340 nm was recorded for 3 min and the change in A 340/min from the initial linear portion of the curve was used to calculate the GPx enzyme activity. The GPx activity was calculated using the extinction coefficient of NADPH (6220 M−1 cm−1) and expressed as units/mg of protein.
Determination of protein carbonyls
The amounts of protein carbonyls in each sample were determined using a modified method by Levine et al. 60 Each of the tissue samples was homogenized in a serine borate buffer (pH 7.4) and then centrifuged at 10,000g for 15 min. Next, 200 μl of supernatant containing approximately 1.5 mg of protein were added to 800 μl of 10 mM 2,4-dinitrophenylhydrazine dissolved in 2 M HCl. In parallel, 200 μl of supernatant were added to 800 μl of 2 M HCl to act as a control. The resulting solutions were incubated at room temperature in the dark for 1 h, with vortexing every 10 min. Subsequently, 1 ml of 20% TCA was added to each solution, vortexed, and then placed on ice for 5 min. The solutions were then centrifuged for 10 min at 10,000g. The resulting pellet was washed three times with an ethanol and ethyl acetate (1:1) mixture. The samples were then left to dry for 10 min, after which 800 μl of 6 M guanidine solution (prepared in 20 mM potassium phosphate and adjusted to a pH of 2.3 using trifluoroacetic acid) were added. The solutions were vortexed to resuspend the proteins. The solutions were centrifuged at 10,000g for 10 min and the supernatant was analyzed by spectrometry. Absorption was measured at a wavelength of 370 nm against the sample blank, and the protein carbonyl content was determined using the associated molar absorption coefficient (22,000 M−1 cm−1).
Determination of protein
Protein levels of the tissue samples were measured by the Bradford method. 61 Concentrated Coomassie Blue (Bio-Rad, Hercules, California) was diluted 1:5 (v/v) with distilled water. Twenty microliters of the diluted tissue homogenate were then added to 1.5 ml of this diluted dye and absorbance was measured at 595 nm using a ultraviolet (UV) spectrophotometer (Shimadzu Scientific Instruments, Columbia, Maryland). Bovine serum albumin was used as the protein standard.
Statistical analysis
All reported values were represented as mean ± SD of quadruplets. Statistical analysis was performed using the GraphPad Prism software (GraphPad, San Diego, California). Statistical significance was ascertained by one-way analysis of variance, followed by Tukey’s multiple comparison tests. Values of p < 0.05 were considered significant.
Results
Effect of METH and NACA on GSH levels in different organs
Compared to the control and the NACA-only treated groups, the METH-treated animals had significantly lower GSH levels in the organs studied. Kidneys, livers, and brains showed a decrease in their GSH levels of approximately 64%, 54%, and 40%, respectively. However, pretreatment of the animals with NACA increased the GSH levels significantly in the METH-exposed animals, indicating that NACA was conferring protection to these animals (Figure 1). Treatment with NACA alone was not found to have a significant impact on the intracellular levels of GSH in all tissues, as compared to the control group, except in the kidneys where an increase in the GSH levels was observed.

Effect of methamphetamine (METH) and N-acetylcysteine amide (NACA) on glutathione (GSH) levels in (A) kidneys, (B) livers, and (C) brains of CD-1 mice. CD-1 mice were injected (intraperitoneally (i.p)) with either saline, for control and METH group, or NACA (250 mg/kg body weight) for the NACA and NACA + METH groups, 30 min before exposure to METH (10 mg/kg body weight). This was followed by four more injections of METH every 2 h over an 8-h period. Mice were killed 24 h after the last METH injection and the GSH levels were measured in homogenized tissue samples. All experiments were performed in quadruplet, and the values reported are mean ± SD (a: different from control group, b: different from METH group, c: different from NACA group; p < 0.05).
Effect of METH and NACA on lipid peroxidation in different organs
Lipid peroxidation is an important consequence of oxidative stress, and can be estimated by measuring the levels of MDA, a stable by-product of lipid peroxidation. METH-exposed animals had about a 22–42% increase in the MDA levels in the organs studied, as compared to the levels in the controls (Figure 2). The kidneys were seen to be affected the most, with about a 42% increase in the MDA levels. The animals in the NACA + METH group experienced significantly less lipid peroxidation than the animals did in the METH-only treated group. Treatment with NACA only did not alter the MDA levels significantly, as compared to those of the control group.
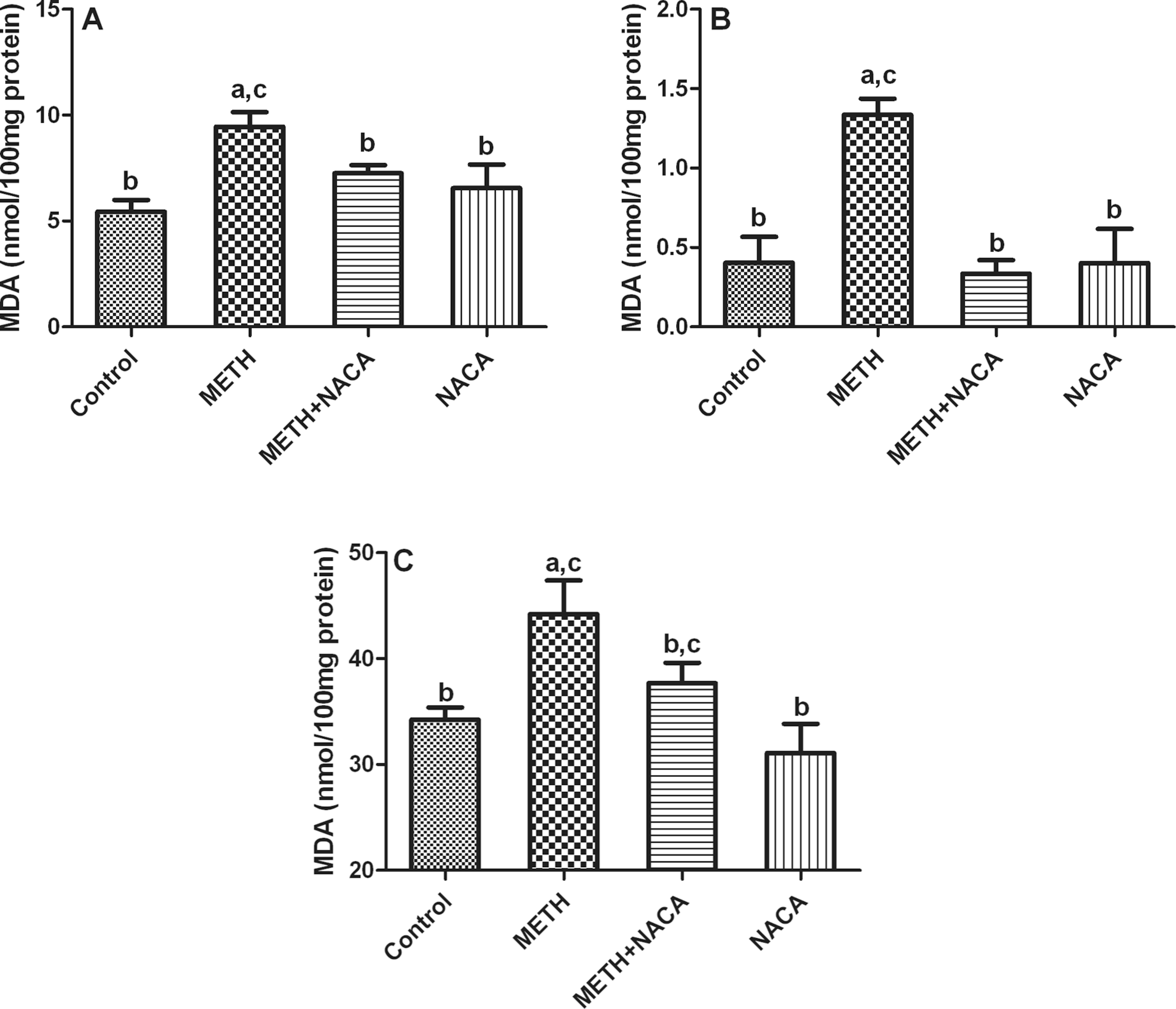
Effect of methamphetamine (METH) and N-acetylcysteine amide (NACA) on lipid peroxidation in (A) kidneys, (B) livers, and (C) brains of CD-1 mice. CD-1 mice were injected (intraperitoneally (i.p.)) with either saline, for control and METH group, or NACA (250 mg/kg body weight) for the NACA and NACA + METH groups, 30 min before exposure to METH (10 mg/kg body weight). This was followed by four more injections of METH every 2 h over an 8-h period. Mice were killed 24 h after the last METH injection and malondialdehyde (MDA) levels were measured in homogenized tissue samples. All experiments were performed in quadruplet, and the values reported are mean ± SD (a: different from control group, b: different from METH group, c: different from NACA group; p < 0.05).
Effects of METH and NACA on protein carbonyl levels
Animals in the METH-only treated group had a 1.5- to 2.0-fold increase in protein carbonyl levels in the organs studied, when compared to the control group. Levels of protein carbonyls in the NACA-only treated group were not significantly different from control levels. However, when animals were pretreated with NACA before exposure to METH, protein carbonyl levels within the organs were significantly lower than those in the METH-treated group (Figure 3).

Effect of methamphetamine (METH) and N-acetylcysteine amide (NACA) on protein carbonyl levels in (A) kidneys, (B) livers, and (C) brains of CD-1 mice. Protein carbonyl levels were measured in homogenized tissue samples for control, METH-only, NACA-only, and METH + NACA groups. The data show that protein oxidation is significantly increased in the tissues of mice exposed to METH. Pretreatment with NACA significantly reduced the levels of protein carbonyls in mice also treated with METH to levels similar to that of the control group. All experiments were performed in quadruplet, and the values reported are mean ± SD (a: different from control group, b: different from METH group, c: different from NACA group, p < 0.05).
Effect of METH and NACA on antioxidant enzyme activity
Effect on GPx activity
Table 1 shows GPx activity in different organs of METH-treated animals. METH administration markedly decreased GPx activity to ∼48–67% of that of the control. Pretreatment with NACA significantly attenuated this decrease in the activity of GPx in the METH-treated animals. NACA alone did not significantly alter the results from those of the control group.
Effects of NACA on the activities of GPx (mU/mg protein)a

GPx: glutathione peroxidase; METH: methamphetamine; NACA: N-acetylcysteine amide.
a All experiments were performed in quadruplet, and the values reported are mean ± SD.
b Significantly different from the METH group at p < 0.05.
c Significantly different from the control group at p < 0.05.
d Significantly different from the NACA group at p < 0.05.
Effect on CAT activity
Antioxidant enzymes like CAT are involved in detoxification of peroxides in the body. Exposure of animals to METH increased CAT activity in the organs studied (∼42–67% increase). CAT activity in the kidneys (∼67%) increased more than in the livers (42%) of METH-treated animals (Table 2). However, reversal in the CAT activity was observed in METH-exposed animals that had been pretreated with NACA.
Effects of NACA on the activities of catalase (mU/mg protein)a

GPx: glutathione peroxidase; METH: methamphetamine; NACA: N-acetylcysteine amide.
a All experiments were performed in quadruplet, and the values reported are mean ± SD.
b Significantly different from the METH group at p < 0.05.
c Significantly different from the control group at p < 0.05.
d Significantly different from the NACA group at p < 0.05.
Discussion
METH abuse is quite prevalent worldwide due to its euphoric effects, wide availability, and relatively low cost. An acute high dose of METH causes serious consequences, including rhabdomyolysis, hyperthermia, renal and liver failure, cardiac arrhythmias, heart attacks, cerebrovascular hemorrhages, strokes, and seizures. 22 –24 Multiorgan dysfunction is seen in acute overdoses, but a vast majority of human METH abusers do not suffer from these complications. Chronic abuse of METH contributes to anxiety, depression, aggressiveness, social isolation, psychosis, mood disturbances, and psychomotor dysfunction. 23,25,26
Oxidative stress is believed to play a crucial role in METH-induced toxicity in the brain and other tissues, as evidenced by findings in previous studies. 47,48,62,63 In the case of the brain, it is believed that, initially, METH causes a massive release of dopamine by inhibiting monoamine oxidase activity and dopamine uptake. 64 With higher doses, however, it causes dopamine depletion by degenerating dopaminergic terminals, damaging dopaminergic neurons and decreasing dopamine transporter numbers. 65 –67 Dopamine then reacts with molecular oxygen to form ROS, such as H2O2, superoxide, and hydroxyl free radicals, resulting in a condition known as oxidative stress 67 and causes neuronal death by apoptosis. 68 Although the precise mechanisms by which METH elicits adverse effects in various organs are unclear, studies have indicated the role of sigma receptors in mediating METH effects. These receptors are a unique group of drug-binding sites that are present in various organs, including the brain, heart, and lungs. 69 Activation of these receptors by METH in peripheral organs may be involved in METH-induced peripheral organ damage. Several studies have indicated that cytotoxicity via mitochondrial dysfunction, resulting in the depletion of intracellular adenosine triphosphate (ATP) as well as adenine nucleotide pools, is a critical factor in the pathologic effect of chemically induced toxicity. 70,71 Ogata et al. have reported that incubation of hepatocytes with amphetamine-derived designer drugs caused cell death accompanied by losses of intracellular ATP, adenine nucleotides, GSH, and mitochondrial ▵Ψ, and an increase in ROS and oxidized glutathione (GSSG) levels, indicating that these drugs induce oxidative stress in isolated rat hepatocytes. 72 Oxidative stress was also implicated in d-amphetamine-induced hepatotoxicity by Carvalho et al. 17 Kubo et al. have implicated oxidative damage in the kidneys of METH abusers. Immunohistochemical analysis revealed the presence of 8-hydroxydeoxyguanosine, 4-hydroxynonenal, and SOD, which are oxidative stress markers in the kidneys of METH abusers. 73
Although oxidative stress is believed to play a key role in METH-induced multiorgan dysfunction, a comparison of the oxidative effects of METH in different organs has not been sufficiently studied. Herein, we report comparable in vivo oxidative effects of METH on the brain, liver, and kidney tissues of CD-1 mice. To the best of our knowledge, we are the first to study and evaluate the potential of a novel thiol antioxidant, NACA, to prevent METH-induced oxidative stress damage to these organs. Preliminary studies in our laboratory demonstrated that NACA was significantly more effective than NAC in providing thiols in the plasma and liver tissue of Wistar rats, which were administered either NACA or NAC, using i.p. injections of 500 mg/kg of each compound. Results indicated that NACA prevented oxidative damage, possibly by scavenging pre-existing ROS, while halting the production of further ROS, lipid peroxidation, and protein carbonylation. In addition, NACA acted by increasing the levels of GSH, as well as the activity of the detoxification enzyme, GPx.
METH-exposed animals showed significant decreases in GSH in all the organs tested, indicating that exposure to METH induced oxidative stress in these organs. However, NACA was capable of restoring GSH levels in these organs (Figure 1). Significant increases in the GSH levels in the kidney tissues in the NACA-only treated group could be attributed to the fact that the activity of γ-glutamyl-cysteine synthetase in this tissue is much higher than that in the liver and brain tissues. 74,75 NACA might be providing cysteine as a substrate for this highly active enzyme in the kidney and thereby significantly increasing GSH levels, even in the absence of any oxidative stress. Our results are in line with the results previously reported by Achat-Mendes et al., 76 Açikgöz et al., 77 and Kim et al. 78 In addition, Moszczynska et al. 38 reported a modest decrease in total GSH concentration after METH administration. However, Flora et al. 79 and Harold et al. 37 reported increases in the total GSH levels immediately after METH administration, possibly due to the cell’s initial defense mechanism against oxidative stress, as the levels were measured within a few hours of METH administration. Increases in the GSH content can be explained by an upregulation of γ-glutamyl-cysteine synthetase in response to oxidative stress. In addition, total GSH (GSH + GSSG) levels may not reflect individual changes in GSH and GSSG. We believe that reduced GSH is a better indicator of oxidative stress; therefore, we determined the reduced GSH levels in the aforementioned organs.
A possible explanation for a decrease in GSH levels is the reduced activity of the enzymes involved in GSH synthesis and/or the oxidation of GSH to GSSG under oxidative stress. An alternative explanation could be an increase in extracellular levels of glutamate which, in turn, might inhibit the uptake of cystine via the glutamate–cystine exchange system, thereby reducing GSH synthesis. The protective effect of NACA is probably mediated by the ability of the NACA to increase GSH biosynthesis by reducing extracellular cystine to cysteine, and/or by supplying the sulfydryl groups that can stimulate GSH biosynthesis, or by conversion of GSSG to GSH by nonenzymatic thiol disulfide exchange. 80
Free radicals attack lipids, especially polyunsaturated fatty acids, in the cell membranes and lead to the formation of by-products like MDA. 81 Our studies indicate that exposure to METH induced a significant increase in MDA levels in various organs of these animals, as compared to the NACA-treated group, thereby pointing to the role of this antioxidant in protecting animals from METH-induced damage. Our results are in accordance with the previously reported increases in lipid peroxidation in response to METH. 42,45 –47,79,82,83 Concomitant reduction in GSH levels (a substrate for GPx) might have hampered the decomposition of lipid peroxides in METH-treated animals. NACA was able to prevent lipid peroxidation by supplying an adequate amount of GSH as a substrate for GPx to effectively decompose lipid peroxides in the mice, thereby reducing MDA levels.
It is well established that proteins, like lipids and DNA, are oxidized by ROS. Oxidative damage to tissue proteins is an important factor in METH-induced organ toxicities. Increased protein carbonylation is a reliable parameter of oxidative stress, as it lacks interference from other nonprotein substances 60 and measures carbonylation of various protein residues, including lysine and arginine. 84 Our results showed that protein carbonyls significantly increased in METH-treated animals, but such increases were prevented by NACA (Figure 3). These results are in line with those reported by Gluck et al. 46
Antioxidant enzymes are involved in the detoxification of lethal peroxides inside the cells. A significant reduction in the activity of GPx, observed after METH administration, may have been partially due to diminished GSH levels that GPx needs as a substrate. We previously reported decreased GPx activity, due to METH, in human brain microvascular endothelial cell culture. 36 Our results are also in accordance with the previously reported decrease in GPx activity after METH administration in brain tissue. 40,42,78 In contrast, the increased GPx activity in rat brains, observed by Flora et al. 79 after METH administration, could have been due to the enhanced levels of cellular GSH observed in their study. On the other hand, no change in GPX activity in rat brains was reported by Moszczynska et al. 38 in response to METH.
Increased CAT activity was observed in animals injected with METH. NACA pretreatment, yet again, reversed CAT activity in the METH + NACA group. Changes in CAT activity had been observed in various tissues that were undergoing oxidative stress. Increased CAT activity in METH-treated animals could be an adaptive response to the higher levels of H2O2 generated by inhibition of GPx. D’Almeida et al. 40 and Melo et al. 47 reported no change in CAT activity when exposed to METH, whereas Jayanthi et al. 42 observed a decrease in CAT activity after METH exposure. We believe that the response of this antioxidant enzyme to oxidative agents is tissue/organ specific and has an adaptive character. Increased CAT activity in METH-treated animals could be an adaptive response to the higher levels of H2O2 generated by the inhibition of GPx. The possible mechanism for the restored CAT activity in METH-exposed mice, when treated with NACA, may be the scavenging of free radicals by NACA or by providing more GSH, which is a substrate for GPx. However, further investigation is needed to confirm this theory.
Depletion of GSH by METH initiates a myriad of events. There is an initial increase in free radicals that overwhelms the scavenging ability of the GSH-dependent enzymes (GPx), which leads to the oxidation of lipids and proteins. Rivière et al. 85 studied the disposition of METH and its metabolite amphetamine and reported the rank order of (+)-METH tissue accumulation after intravenous dosing as kidney > spleen > brain > liver > heart > serum, with terminal elimination half-life values ranging from 53 to 66 min. In another study, Volkow et al. 86 studied METH distribution and accumulation in the human body via positron emission tomography and reported that the liver, brain, and kidney had relatively higher uptakes of the drug. It was distributed through most organs. They reported that the order of uptake was lungs > liver > brain > kidneys. METH’s clearance was fastest in the heart and lungs; slowest in the brain, liver, and stomach; and intermediate in the kidneys, spleen, and pancreas. Our results indicated maximum oxidative stress in the kidneys followed by the liver and brain.
Despite different results reported by the two groups, a high accumulation of METH in most body organs is likely to contribute to the medical complications associated with METH abuse and, therefore, treatment of METH abusers should focus on multiorgan damage. The multiple roles of NACA in preventing METH-induced toxicity include scavenging of free radicals (directly and indirectly) by providing GSH, and chelating Cu2+, ultimately preventing catalyzation of ROS formation, providing cysteine for GSH synthesis, nonenzymatic reduction of the preexisting toxic GSSG into GSH, and stimulating cytosolic enzymes involved in GSH regeneration. Studies have shown the protective effects of NACA against radiation-induced toxicity, human immunodeficiency virus (HIV) protein-induced oxidative stress, allergic airway disease, pollutant-induced lung disease, and lead-induced cell death. 51,52,87 –89
We believe that NACA also prevents METH-induced oxidative stress by scavenging free radicals. This hypothesis is supported by studies in which edaravone (3-methyl-1-phenyl-2-pyrazolin-5-one), a potent scavenger of hydroxyl radicals, prevented METH-induced neurotoxicity.
90,91
This is also supported by studies in which antioxidants like NAC,
Currently, no medications are available to safely reverse life-threatening METH overdoses quickly. Medications that could help METH users recover rapidly from the toxic effects of chronic use are highly desirable for compliance to the current treatment regimen. Findings from this study will aid in developing potentially effective adjunctive therapy with existing METH treatment for addressing oxidative stress-induced organ damage and accompanying psychiatric problems. Based on the encouraging results from the use of NAC and other antioxidants, we believe that NACA would be more effective in treating METH-induced toxicities because of its better permeability across membranes and low toxicity profile. Combining NACA with current treatment medications, as a neuro- and tissue-protective agent, might be an effective approach to treating METH abusers. Additional studies are needed to further delineate and clarify strategies that might improve the treatment of METH-induced toxicity. In any case, our future studies will focus on the antioxidant effects of NACA on acute METH intoxication.
NACA might also be beneficial in reducing drug cravings. These cravings are believed to be caused by glutamate dysfunction within the accumbens nucleus. 97 A mixed disulfide exchange via NACA can result in the formation of cystine, which would help restore the extracellular glutamate concentration in the accumbens nucleus by stimulating the inhibitory metabotropic glutamate receptors and, thereby, reducing synaptic release of glutamate. 97 –99 On the other hand, a recent study of NAC plus naltrexone did not show significant improvement in the reduction of cravings. 100
In summary, data from the present study indicated oxidative stress in the livers, kidneys, and brains of METH-treated animals, with the kidneys being affected the most. Administration of NACA, however, resulted in a significant reduction in the oxidative stress induced by METH, thereby suggesting a therapeutic potential for this novel antioxidant as a tissue- and neuro-protectant against METH toxicity. Further investigation could help determine NACA’s efficacy in METH-abuse treatment and an evaluation of the histology of these organs in METH-treated animals would provide additional information on its therapeutic potential.
Footnotes
Acknowledgments
The authors appreciate the editorial efforts of Barbara Harris and Youyou Zheng.
Funding
This work was supported by the NIDA, NIH (grant number 1 R15DA023409-01A2).