
Abstract
Classically activated macrophages (M1) are proinflammatory effectors and closely related to the progression of neurotoxicity. As a powerful psychostimulant and addictive drug, methamphetamine (Meth) abuse could result in long-lasting abnormalities in retina. This study investigated the effect of Meth at nontoxic concentration on macrophage activation state and its resultant toxicity to photoreceptor cells. Results showed that cytotoxicity was caused by Meth on 661 W cells after coculturing with RAW264.7 macrophage. RAW264.7 cells tended to switch to the M1 phenotype, releasing more proinflammatory cytokines after treatment with Meth. Meth could also upregulate the M1-related gene and protein expression. Our study demonstrated that Meth promoted macrophage polarization from M0 to M1 and induced inflammatory response, providing the scientific rationale for the photoreceptor cell damage caused by the Meth abuse.
Introduction
Methamphetamine (Meth) is an illicit and potent psychostimulant drug that could easily cross the blood–brain barrier with wide-ranging effects on the central nervous system by causing brain damage and neuropsychiatric disorders. 1,2 Millions of individuals used amphetamine-type stimulants worldwide every year, predominantly Meth. 3 Acutely, Meth intake produces hyperactivity, euphoria, increased vigilance, and cardiovascular changes. 4,5 Despite the well-known effects of Meth in different brain areas, only a few studies explored the effects of Meth on visual system, specifically the retina. 6,7 Chronic exposure or taken in binge doses, Meth can cause irreversible damage to photoreceptor cells, resulting in retina degeneration. 8 Studies showed Meth damages monoamine-containing nerve terminals and generates an imbalance in the release and reuptake of dopamine and norepinephrine, leading to neurological abnormalities and eventually psychiatric disorders. 9,10
Meth caused toxicity in retina via multiple mechanisms. Repeated treatment with Meth to rats caused imbalance of oxidative/antioxidative system in retina, and the compromised antioxidant defense may interfere with the normal functioning of the visual system and play a role in the pathophysiology of ocular degenerative events that are linked to increased oxidative stress. 8 The electroretinogram were altered by administration of dopamine agonists, antagonists, or depletion of retinal dopamine levels, suggesting that any substance, such as Meth, which changes the dopaminergic system of the retina would influence vision. 11 –14 Meth at high dose results in direct toxicity; however, low dose of Meth could not affect the viability of cultured photoreceptor cells but caused toxicity in animals after long-term treatment, 15,16 indicating that Meth induces toxicity through other potential pathways. Neuroinflammation has been implicated as an additional mechanism relating with several neurological disorders. 17 It has been well established that Meth exposure activates microglia and astrocytes, which may contribute to Meth-induced neurotoxicity, as activated astrocytes or microglia produce reactive oxygen species and proinflammatory cytokines to propagate the neuroinflammatory cascade to amplify the neuron damage. 18,19
Activated M1-macrophages produce various cytokines and exhibit diverse functions, including proinflammatory responses, tissue destruction, and tumoricidal function, which are closely related to the polarized activation state. 20 This study was performed to explore the effect of Meth on macrophage polarization and the resultant effects on photoreceptor cells.
Materials and Methods
Cell Line, Chemicals, and Reagents
Mouse photoreceptor-derived 661 W cell line and mouse macrophage RAW264.7 cell lines were obtained from Shanghai Cell bank (Shanghai, China). Meth was purchased from National Institutes for Food and Drug Control (Beijing, China). Fetal bovine serum (FBS) and Dulbecco Modified Eagle medium (DMEM) were purchased from Gibco BRL (Gaithersburg, Maryland, USA). 3-(4,5-dimethylthiazol)-2,5-diphenyltetrazolium-bromid (MTT) was purchased from Sigma-Aldrich (St. Louis, Missouri, USA). Lactate dehydrogenase (LDH), nitric oxide (NO), interleukin (IL)-1β, IL-6, IL-12, and tumor necrosis factor α (TNFα) assay kits were purchased from Jiancheng Biological Engineering (Nanjing, China). Cytochrome C enzyme-linked immunosorbent assay kit was from R&D systems (Minneapolis, Minnesota). Cell death detection ELISAplus kit was from Roche Applied Sciences (Switzerland). 5,5′,6,6′-tetrachloro-1,1′,3,3′-tetraethylbenzimidazolylcarbocyanine iodide (JC-1) were purchased from Molecular Probes (California). Caspase-3 and caspase-9 activity assay kits were purchased from Kaiji Institute of Biological Engineering (Nanjing, China). Interferon γ (IFN-γ ) and LPS were purchased from Sigma-Aldrich (St. Louis, Missouri). Real-time polymerase chain reaction (RT-PCR) reagents were purchased from Takara Bio Inc. (Shiga, Japan). All solvents and chemicals used in this study were of analytical grade and purchased from Sinopharm Chemical Reagent Co. Ltd. (Shanghai, China).
Cell Culture and Treatment
661 W cells or macrophage RAW264.7 cells were cultured in DMEM, supplemented with 10% FBS, 100 U/mL penicillin, and 100 U/mL streptomycin at 37°C in a humidified atmosphere of 95% air and 5% CO2. 661 W or RAW264.7 cells were treated with Meth at different concentrations (5, 10, and 20 µmol/L) for 48 hours. 661 W and RAW264.7 cells were cocultured using Transwell 6-well plates (Corning, New York, USA) to study their interaction. Cocultured 661 W and RAW264.7 cells were treated with Meth at 20 µmol/L for 48 hours. RAW264.7 cells were treated with Meth at 20 µmol/L for 48 hours. LPS (100 ng/mL)/INF-γ (10 ng/mL) was used as the positive control.
Cell Viability Measurement
Cell viability was determined by MTT assay. After treatment, cells were plated in 96-well plates at a density of 4 × 104/100 µL. About 10 µL of MTT solution at 0.5 mg/mL was added and incubated for 4 hours at 37°C. The formazan crystals were solubilized with 100 µL dimethyl sulfoxide, and the absorbance was measured at 570 nm by a microplate reader. Cell viability was expressed as the percentage of the negative control, which was set to 100%.
LDH Leakage Assay
LDH assay kit was used to measure the LDH leakage. After treatment, the medium was collected and centrifuged at 3000 rpm for 10 minutes at 4°C. The activity of LDH was measured according to the manufacturer’s instructions. The absorbance was measured by a microplate reader at 490 nm. The LDH leakage content was expressed as the percentage of the negative control, which was set to 100%.
Measurement of Mitochondrial Membrane Potential
The mitochondrial membrane potential (MMP) was measured by the fluorescent probe JC-1. The ratio between green (monomer) and red (aggregate) fluorescence can reflect the MMP. After treatment, 661 W cells were washed with phosphate-buffered saline (PBS) and incubated with JC-1 for 15 minutes at 37°C in the dark. The fluorescence intensity of the red/green was determined by a fluorescence microplate reader (TECAN Polarion, United Kingdom) at an excitation of 490 nm and emission of 530 nm (green fluorescent) and 590 nm (red fluorescent), respectively. The change in MMP was expressed as a percentage of the negative control, which was set to 100%. 21
Cytochrome C Assay
The cytosolic cytochrome C was measured by the assay kit according to the manufacturer’s instructions. After treatment, 661 W cells were washed with PBS and fractionated. After reaction, the optical density was measured by a microplate reader at 490 nm.
Measurement of DNA Fragmentation
DNA fragmentation quantification was measured by Cell Death Detection ELISAplus kit. After treatment, 661 W cells were washed with PBS and lyzed. After a centrifugation at 1000 rpm at 4°C for 10 minutes, 20 µL of supernatant was transferred to a streptavidin-coated microplate and incubated with a mixture of anti-histone–biotin and anti-DNA peroxidase. The peroxidase amount in the immunocomplex was quantified by adding 3-ethylbenzthiazoline-6-sulfonic acid as the substrate. The absorbance of the reaction mixture was measured by a microplate reader at 405 nm. The extent of DNA fragmentation was expressed as a percentage of the negative control, which was set to 100%. 21
Measurement of Caspase Activity
The caspase activity was measured by the assay kit according to the manufacturer’s instructions. After treatment, 661 W cells were washed twice and lyzed. Cell extracts were centrifuged at 20 000 rpm at 4°C for 10 minutes, and aliquots of supernatants containing 25 µg protein were added to a reaction buffer. The reactions were initiated after addition of the fluorescent substrates (50 mmol/L final concentration): Ac-DEVD-Amc for caspase-3 activity and Ac-LEDH-Afc for caspase-9 activity. After incubation at 37°C for 2 hours, the cleavage of the substrates was measured (Amc: 390/475 nm; Afc: 400/505 nm) by a microplate reader. The activity of caspase was expressed as a percentage of the negative control, which was set to 100%. 21
Cytokine Assays
After treatment, supernatant of culture medium was collected to determine the cytokine levels. NO was determined by Griess assay according to the manufacturer’s instructions. IL-1β, IL-6, IL-12, and TNFα levels were detected using murine ELISA kits according to the manufacturer’s instructions.
Real-Time PCR
After treatment, 661 W cells were collected to extract the total RNA. Complementary DNA was generated by adding 1 µg of the total RNA to SuperScript master mix and performing reverse transcription. Quantitative PCR was performed using SYBR Green Supermix (Bio-Rad, California). Comparative C t value method was used to quantify the expression of genes of interest in different samples. The messenger RNA (mRNA) levels were normalized to a housekeeping gene Gapdh.
Western Blot Analysis
After treatment, cells were harvested and washed with PBS. Protein was isolated and quantified using the protein assay kit (Biyotime, China). Cell lysates in 5× sodium dodecyl sulfate (SDS) sample buffer were boiled for 5 minutes and then equal amounts of protein (40 μg) were subjected to electrophoresis on a 12% (v/v) SDS-polyacrylamide gel. After proteins were electroblotted to a polyvinyl difluoride membrane, the membrane was blocked with PBS containing 2% BSA at room temperature and incubated with indicated primary antibodies at 4°C overnight, followed by incubating with the appropriate horseradish peroxidase-conjugated secondary antibody for 1 hour. After incubation, membrane was washed 3 times, and the antigen–antibody complexes were visualized by the enhanced chemiluminescence system (PerkinElmer, MA, USA).
Statistical Analysis
Values were presented as mean ± standard deviation and analyzed by 1-way analysis of variance using the Sigma Stat statistical software (SPSS Inc., Chicago, Illinois). Differences were considered as significant at P < .05.
Results
Effects of Meth on RAW264.7 and 661W Cell Viability
RAW264.7 or 661 W cells were treated with Meth at different concentrations. Comparing to the negative control, no significant difference in the optical density value was observed, indicating 661 W or RAW264.7 cell viability was not significantly reduced after treatment with Meth, as shown by Figure 1.
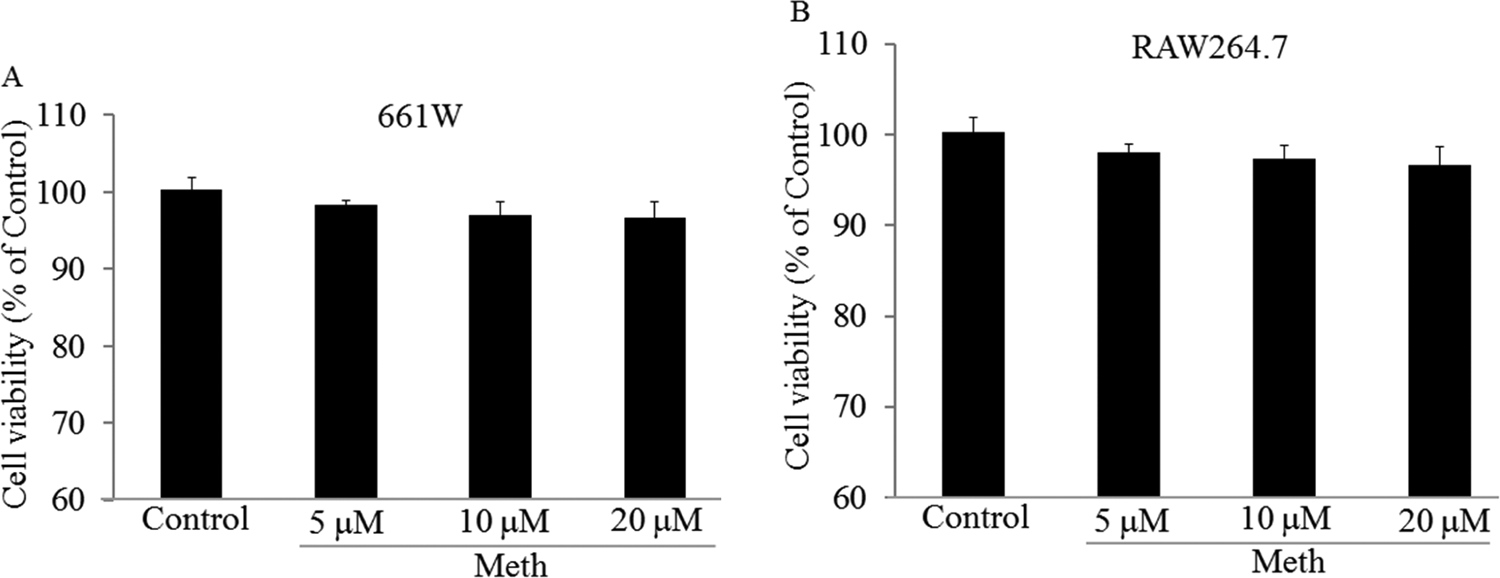
661 W or RAW264.7 cells were treated with Methamphetamine (Meth) at 5, 10, and 20 µmol/L for 48 hours and then tested the cell viability by MTT assay. Meth did not cause significant toxicity on 661 W (A) and RAW264.7 (B) cells. Samples were measured in triplicate and experiments were repeated 3 times.
Meth Caused Toxicity on 661 W Cells Through Coculturing With RAW264.7 Cells
Meth at 5, 10, and 20 µmol/L did not cause toxicity on 661W cells. However, after coculturing with RAW264.7 cells, significant toxicity on 661W cells caused by Meth was observed (Figure 2A), together with the increasing leakage of LDH (Figure 2B), decreasing of MMP (Figure 2C), increasing of the cytochrome C releasing into cytoplasm (Figure 2D), increasing DNA fragmentation in 661 W cells (Figure 2E), increasing caspase activities (Figure 2F), and changing apoptosis-related protein (Figure 2G; P < .05).
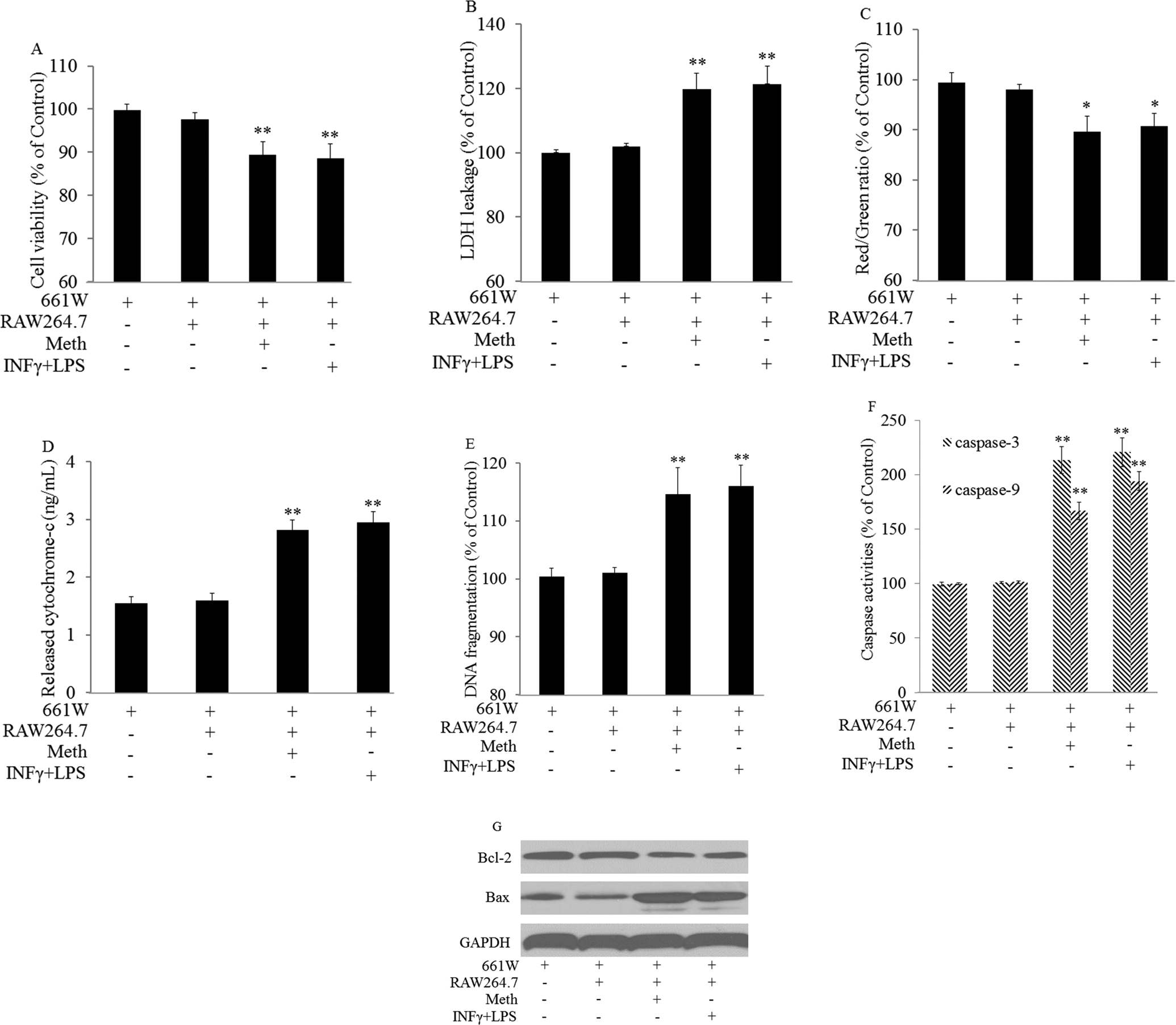
Methamphetamine (Meth) caused cell damage on 661 W cells after coculturing with RAW264.7 cells. 661 W and RAW264.7 cells were cocultured using Transwell plates and treated with Meth at 20 µmol/L for 48 hours. Cocultured cells were treated with IFNγ (10 ng/mL)/LPS (100 ng/mL) as the positive control. 661 W cells were collected to test the cytotoxicity. Cytotoxicity were determined by: decreasing cell viability (A); increasing LDH leakage (B); decreasing mitochondrial membrane potential (C); increasing cytochrome-c releasing (D); increasing DNA fragmentation (E); increasing caspase activities (F); and changing apoptosis-related protein expression (G). *P < .05, **P < .01 versus 661 W cells alone. Samples were measured in triplicate and experiments were repeated 3 times.
Meth Increased the IL-1β, IL-6, IL-12, TNFα Levels and NO Production in RAW264.7 Cell
RAW264.7 cells were treated with Meth at 20 µmol/L for 48 hours. Comparing to the negative control, Meth treatment significantly increased IL-1β, IL-6, IL-12, TNFα levels, and NO production in the supernatant. The effects were comparable to the positive control (P < .01; Figure 3).
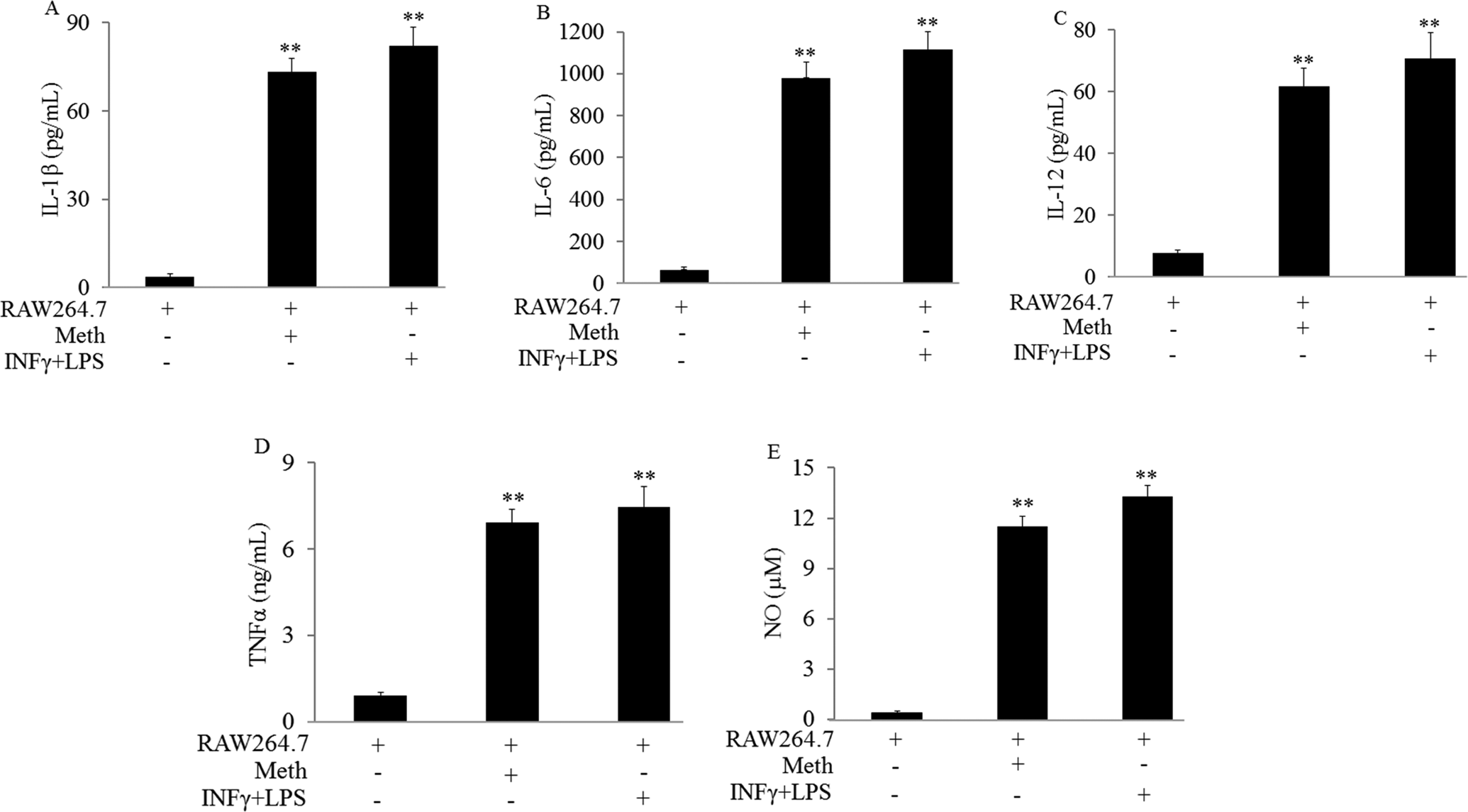
Methamphetamine (Meth) promoted RAW264.7 cell polarization from M0 to M1. RAW264.7 cells were treated with Meth at 20 µmol/L for 48 hours. RAW264.7 cells were treated with IFNγ (10 ng/mL)/LPS (100 ng/mL) as the positive control. Macrophage polarization was determined by M1 markers: A, IL-1β. B, IL-6. C, IL-12. D, TNFα. E, NO. **P < .01 versus RAW264.7 cells alone. Samples were measured in triplicate and experiments were repeated three times.
Meth Increased the mRNA Expression of IL-1β, IL-6, IL-12, and TNFα in RAW264.7 Cell
RAW264.7 cells were treated with Meth at 20 µmol/L for 48 hours. Meth treatment significantly increased mRNA expression of IL-1β, IL-6, IL-12, and TNFα (P < .01) (Figure 4).
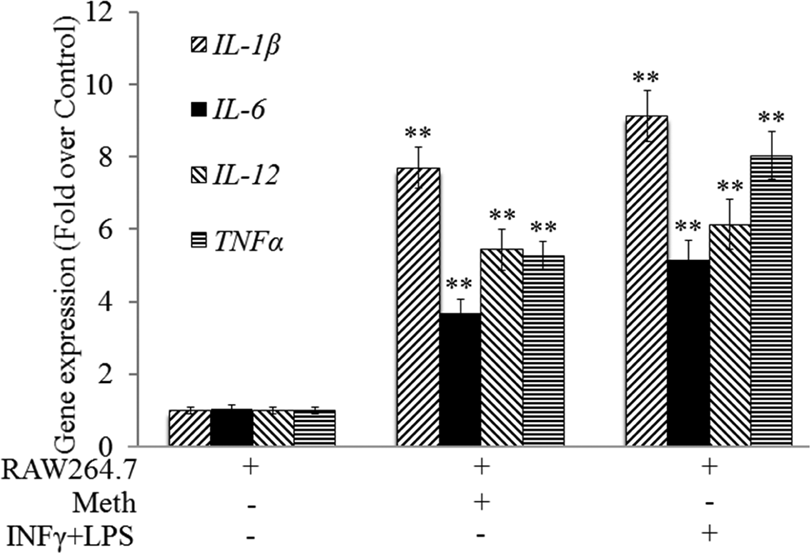
Meth promoted RAW264.7 cell polarization by increasing gene expression. RAW264.7 cells were treated with Methamphetamine (Meth) at 20 µmol/L for 48 hours. RAW264.7 cells were treated with interferon γ (IFNγ; 10 ng/mL)/LPS (100 ng/mL) as the positive control. RAW264.7 cells were collected to determine gene expression. Macrophage polarization was determined by the M1 marker gene expression. **P < .01 versus RAW264.7 cells alone. Samples were measured in triplicate and experiments were repeated 3 times.
Meth Increased the Protein Expression of IL-1β, IL-6, IL-12, and TNFα in RAW264.7 Cell
RAW264.7 cells were treated with Meth at 20 µmol/L for 48 hours. Meth treatment significantly increased protein expression of IL-1β, IL-6, IL-12, and TNFα in RAW264.7 cell (Figure 5).
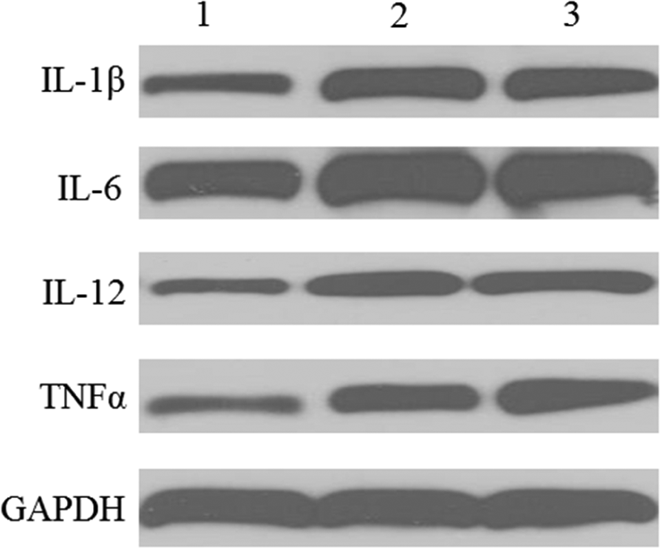
Methamphetamine (Meth) promoted RAW264.7 cell polarization by changing protein expression. RAW264.7 cells were treated with Meth at 20 µmol/L for 48 hours. RAW264.7 cells were treated with interferon γ (IFNγ;10 ng/mL)/LPS (100 ng/mL) as the positive control. RAW264.7 cells were collected to determine protein expression. Lane 1: RAW264.7; Lane 2: RAW264.7+Meth; Lane 3: RAW264.7+ IFNγ/LPS. Experiments were repeated 3 times and a representative blot was displayed.
Discussion
Although much progress has been made to understand the neurological consequences of Meth abuse, there is far less about the effects of Meth on retina. Even little is known about the Meth-induced chronic neurotoxicity, and there are no efficacious therapies for this neural injury. 22,23 This study is the first to explore its underlying mechanism. Different from previous studies that Meth directly caused cytotoxicity through oxidative stress, 8 our results showed that Meth at nontoxic concentration on 661 W cells caused cell damage by first promoting the M1 polarization of macrophages to induce inflammatory responses, instead of rendering cell death directly. Our study provided the other potential pathway and complemented the previous research into the mechanisms underlying retina toxicity caused by Meth exposure.
During the process of cell death regulation, mitochondria play a pivotal role, as many genes and proteins influence the apoptosis progression through the mitochondrial pathway. 24 Mitochondrial membrane potential decreasing would induce release of cytochrome C from the mitochondria to nucleus and activate caspase-related apoptotic protein, resulting in chromatin condensation and DNA cleavage. As a biochemical hallmark, DNA fragmentation has been widely used as apoptosis index. 25,26 Caspase 3 is an apoptotic executor and can activate endonucleases to cleave nuclear DNA, ultimately leading to cell death. Cytochrome C releasing from mitochondria together with caspase 9 precursor led to caspase 9 activation. 21 In the present study, treatment of 661W cells with Meth in the presence of RAW264.7 cells caused cell injury, accompanied by decrease in MMP, cytochrome C release from mitochondria, DNA fragmentation, and increased activities of caspase 3 and caspase 9. Proapoptotic protein of bax expression was increased, and antiapoptotic protein of bcl-2 expression was decreased. These data indicated Meth caused cytotoxicity through affecting the macrophages.
Heterogenic macrophages are divided into 2 subpopulations: classically activated macrophages (M1) and alternatively activated macrophages (M2). 27 The M1 macrophages are defined as potent effector cells exhibiting antimicrobial abilities. IFNγ and LPS activate M1 macrophage, which exhibits high phagocytic capacities with more production of toxic intermediates (eg, reactive oxygen species and NO). On the other hand, IL-4 activates M2 macrophage, promoting tissue repair, angiogenesis, and tumor progression. 28
Macrophages play important roles in the regulation of immune response and inflammation and are involved in various diseases. 29,30 Activated M1 macrophages secrete a lot of inflammatory mediators such as NO, and inflammatory cytokines such as TNFα, IL-1β, IL-6, and so on. 31,32 These molecules are important mediators involved in the progress of many inflammatory diseases.
Meth-induced inflammatory response in different brain areas, including striatum, hippocampus, cerebellum, and frontal cortex. 33 –35 Cytokines and chemokines released by activated microglia appeared to play important roles in Meth-induced neuronal injury and neuropsychiatric impairments, including cognitive deficits, depression, and anxiety. 36 In this study, exposure of RAW264.7 cells to Meth induced M1 polarization, which was characterized by the production of NO, TNFα, IL-1β, IL-6 and IL-12, and relevant genes and proteins. All of these contributed to inflammation and exacerbate cell injury. After Meth treatment, proinflammatory cytokines increased, together with the change in gene and protein expression, suggesting that Meth could enhance macrophage polarization from M0 to M1. Resultantly, Meth promoted inflammatory response to cause cytotoxicity on 661 W cells in the coculturing system.
In conclusion, our study demonstrated that Meth at nontoxic concentration caused toxicity on photoreceptor cells through promoting macrophage polarization and inflammatory response. Our study provided the scientific rationale for the photoreceptor cell damage caused by the chronic use of Meth. Our work also suggests immunotherapeutic strategies that target specific neuroinflammatory pathways may reduce retina toxicity of Meth.
Footnotes
Authors’ Note
Aihui Zhang and Laiwei Wu contributed equally to this study.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.