
Abstract
Autophagy and apoptosis are important players in the progression of hepatic fibrosis via activation of hepatic stellate cells (HSCs). Despite the recently depicted antifibrotic effects of alpha-lipoic acid (ALA), however, its modulatory effects on HSCs autophagy remain unverified. Our study aimed to elucidate the underlying antifibrotic mechanisms through which ALA mediates HSC autophagy and apoptosis. Liver fibrosis was induced via thioacetamide (TAA) intoxication in rats; TAA-intoxicated rats were treated with either silymarin or ALA. Effect of ALA on biochemical parameters and immunohistopathological examinations was measured and compared to silymarin. ALA restored normal hepatic architecture (S1 vs. S4), liver functions, hepatic glutathione, and transforming growth factor-β1 levels. ALA ameliorated hepatic levels of malondialdehyde, platelet-derived growth factor, tissue inhibitor metalloproteinases-1, hydroxyproline, and expression of alpha-smooth muscle actin. Moreover, ALA significantly reduced messenger RNA expression of LC3-II genes and triggered caspase-3 expression. Interestingly, ALA exhibited superior activities over silymarin regarding suppression of proliferation, activation and autophagy of HSCs, collagen deposition, and induction of HSCs apoptosis. In conclusion, treatment of TAA-intoxicated rats with ALA inhibited autophagy and induced apoptotic clearance of activated HSCs. Accordingly, this study provides mechanistic insights into the possible applicability of ALA in the treatment of hepatic fibrosis.
Keywords
Introduction
Liver fibrosis is a serious health condition that results from virtually all forms of liver injury including inherited, metabolic, inflammatory, and infectious diseases, drugs, and toxins. 1 Hepatic fibrosis persists for decades and if left untreated, it progresses into liver cirrhosis, hepatocellular carcinoma, and eventually death. 1,2
Hepatic stellate cells (HSCs) are the key fibrogenic cell type that orchestrates the fibrogenesis process in the injured liver. 3 Their proliferation and activation are pivotal players in the progression of hepatic fibrosis through producing the main extracellular matrix (ECM) components including collagen type I and α-smooth muscle actin (α-SMA) 3 as well as in the release of profibrogenic and promitogenic cytokines. 4 Furthermore, activated HSCs receives paracrine signals from apoptotic hepatocytes, inflammatory cells, and Kupffer cells leading to the production of profibrogenic cytokines and growth factors, which further stimulate HSCs activation and ECM deposition. 5
Several signaling pathways were evidenced to participate in the progression hepatic fibrogenesis and autophagy appears to be one of the critical contributors. 6 Autophagy is an important conserved process needed to maintain intracellular energy balance. 7 Autophagy is strongly associated with liver fibrosis, where the process of HSCs activation is autophagy-dependent to supply the energy required to establish the activated phenotype via degradation of stored lipid droplets. 8 This takes place through lipid mobilization, liberation of free fatty acids, and mitochondrial β-oxidation, which maintain HSCs energy homeostasis. 9 Although, it was evidenced that hepatic fibrosis is a dynamic reversible process, 10 not only in experimental models of liver fibrosis 11 but also in humans 12,13 ; yet the development of antifibrotic therapies remains elusive. 14
Alpha-lipoic acid (ALA, 1,2-dithiolane-3-pentanoic acid), also known as thioctic acid, is a natural dithiol compound synthesized from octanoic acid in the mitochondrion. 15 ALA possesses antioxidant and protective effects against diabetic neuropathy, 16 kidney injury, 17 and retinopathy. 18 ALA also has shown a regulatory role on autophagy in cardiomyocytes, 19 injured retinal pigment epithelial cells, 20 and human umbilical vein endothelial cells. 21 Although, ALA was recently shown to display antifibrotic effects in rats, 22,23 yet its impact on autophagy in HSCs has not been documented.
In this study, we elucidated the antifibrotic potential of ALA against thioacetamide (TAA)-induced liver fibrosis in male rats and we further clarified its underlying protective mechanisms. ALA administration resulted in the blockade of activation and proliferation of activated HSCs, induced their apoptosis, and mitigated collagen deposition within hepatic tissues. We also verified that ALA modulated the progression of hepatic fibrosis via autophagy inhibition, which may limit the available energy for the activation of HSCs.
Materials and methods
Chemicals and reagents
Silymarin (Legalon®) and ALA (Thiotacid®) were purchased from Chemical Industries Development and Eva Pharma, respectively, Cairo, Egypt. All chemicals and solvents were of the highest commercially available grade.
Animals
Adult male Sprague–Dawley rats, weighing 250–300 g, were purchased from the animal house of Theodor Bilharz Research Institute, Giza, Egypt. All rats were housed and maintained under an environmentally controlled room at 20–22°C, 12-h light/ 12-h dark cycle and 50–60% humidity with free access to food and water ad libitum throughout the acclimatization (7 days) and experimental periods. The animal experiments were conducted in accordance with the guide for the care and use of laboratory animals of the National Institutes of Health (1996) with its amendments and were approved by the Institutional Review Board of Theodor Bilharz Research Institute (April 29, 2013).
Experimental design
Thirty-two rats were randomly divided into four groups of eight animals each: group I: served as normal control and received intraperitoneal (ip) injections of saline; group II: rats were given ip injections of TAA in a dose of 200 mg/kg twice a week for 12 weeks. 24 TAA-intoxicated rats in groups III and IV were treated with silymarin (50 mg/kg) 25 and ALA (30 mg/kg), 26 respectively. Drugs were administered once daily for 8 weeks via oral gavage starting from the fifth week post TAA intoxication at an apparent stage of fibrosis (stage 2), which was verified upon histopathological examination of hepatic tissues (Figure 1(a)). Forty-eight hours after the last treatment, rats were killed with an ip injection of ketamine (80 mg/kg), next blood samples and liver tissues were collected for subsequent analyses.

Effect of ALA-treatment daily for 8 weeks on hepatic histopathology and collagen deposition. (a) Histopathological examinations of TAA-intoxicated hepatic sections for 4 weeks stained with H&E (×200) showing hepatic fibrosis of S2 with disorganized hepatic lobular architecture and hepatocytes vacuolar degeneration. (b) Histopathological examinations of hepatic sections stained with H&E (×200) showing livers of control rats with normal hepatic architecture, livers of TAA-intoxicated rats with disturbances in hepatic architecture of S4 (hepatic cirrhosis), livers of TAA-intoxicated rats treated with silymarin displaying recovery of hepatic tissues with mild dilation of sinusoids and lymphocyte infiltration (S2), and livers of TAA-intoxicated rats treated with ALA representing preserved hepatic architecture (S1). (c) Picro-Sirius red stain (×50) where the distribution of collagen fibers (black arrows) was visualized using imaging analysis software. (d) Numerical scoring of fibrosis score. Data are expressed as mean (n = 6) ± SD; *p < 0.05 versus normal control group; † p < 0.05 versus TAA-intoxicated group. TAA: thioacetamide, ALA: α-lipoic acid; SD: standard deviation; H&E: hematoxylin/eosin.
Assessment of hepatotoxicity and oxidative stress markers
Serum ALT and AST were spectrophotometrically determined using the commercially available kits (Spectrum, Cairo, Egypt). Meanwhile, liver tissues were washed, homogenized in saline (pH 7.4), and the supernatants were used for the determination of reduced glutathione (GSH) and lipid peroxidation (malondialdehyde (MDA)) levels according to the methods described by Ellman 27 and Ohkawa et al., 28 respectively.
Assessment of fibrosis markers
The hepatic levels of platelet-derived growth factor (PDGF)-BB, transforming growth factor (TGF)-β1, and tissue inhibitor metalloproteinases (TIMP)-1 were determined in liver tissue homogenates using the commercial enzyme-linked immunosorbent assay kits (Quantikine R&D systems, Minneapolis, Minnesota, USA). Moreover, hepatic hydroxyproline (HP) content was colorimetrically quantified in liver homogenates. 29 Briefly, dried liver tissue was hydrolyzed in 6 N HCl and then dissolved in isopropanol and chloramine-T solution followed by reaction with p-dimethylaminobenzaldehyde. The absorbance of samples was determined at 540 nm and the amount of HP was expressed as mg/g wet tissue.
Determination of autophagy using LC3-II mRNA expression
LC3-II messenger RNA (mRNA) expression was estimated by quantitative real-time polymerase chain reaction (qRT-PCR). For RNA extraction, total RNA from liver tissue was extracted using the QIAzol and RNeasy mini kit (QIAGEN, Valencia, CA, USA), in accordance with the manufacturer’s instructions. Two milligrams of RNA were used to synthesize cDNA (Thermo Scientific, Waltham, Massachusetts, USA), which were then amplified using SYBR Green master mix (Thermo Scientific). RNA samples were then reverse transcribed and processed for PCRs. The mRNA expression of LC3–II was calculated based on the method of 2−(ΔΔ
Ct
), where Ct is cycle threshold; the results were normalized to β-actin. The primers were designed and synthesized (Invitrogen, Waltham, Massachusetts, USA) as follows: LC3-II: Forward: 5′ATGCCGTCCGAGAAGACCTTCAAA3′; reverse: 5′TTACACAGCCAGTGCTGTCCCGA3′ β-actin: Forward: 5′ATTGCTGACAGGATGCAGAA3′; reverse: 5′TAGAGCCACCAATCCACACAG3′
Liver histology and fibrosis grade
Liver specimens, embedded in paraffin blocks, were stained by either hematoxylin/eosin (H&E) (4 mm thickness) or Sirius red (20 mm thickness) for the analyses of overall liver histology and collagen distribution, respectively. Collagen was quantified using imaging analysis software (Axiovision L.E. 4.8; Carl Zeiss MicroImaging, Jena, Germany); the red-stained area (mm2) was measured in five consecutive fields (×50). Moreover, a numerical scoring system for morphometric analysis of hepatic fibrosis score was quantified. 30 Briefly, healthy liver was classified as stage (0): no fibrosis; stage (1): expansion of fibrosis in portal area localized in perisinusoidal and intralobular fibrosis; stage (2): peripheral fibrosis in portal area, formation of fibrous septum, and retention of intralobular architecture; stage (3): fibrous septum accompanied by intralobular structural disorders and no hepatic cirrhosis; and stage (4): early hepatic cirrhosis characterized by bridging fibrosis with pseudolobular formation.
Immunohistochemical parameters
Hepatic sections were immunohistochemically stained for caspase-3 and α-SMA with a horseradish-peroxidase complex kit (Abcam Inc., Cambridge, UK). The percent of positively stained brown cytoplasm (α-SMA and caspase-3) were examined in 10 microscopic fields (at ×400 under Zeiss light microscopy, Jena, Germany).
Statistical analysis
Data are expressed as mean ± standard deviation. Statistical analysis was performed using one-way analysis of variance test followed by Tukey’s post hoc test for multiple comparisons. The difference in fibrosis stages between groups was analyzed using the nonparametric Kruskal–Wallis test followed by Dunn procedure (version 5.03; GraphPad Software, San Diego, California, USA). Differences were considered significant when p values <0.05.
Results
ALA protected rat liver from TAA-induced hepatic injury and fibrogenesis
Serum levels of ALT and AST were measured to evaluate the effect of ALA administration on TAA-induced liver injury. As presented in Table 1, the activities of serum ALT and AST were significantly elevated in TAA-intoxicated rats by 98.84% and 38.61%, respectively, when compared to their corresponding normal controls. Treatment with silymarin significantly improved ALT and AST levels by 30.52% and 16.20%, respectively, when compared to their corresponding TAA-intoxicated groups; while ALA effectively restored their normal levels.
Effect of ALA-treatment on TAA-induced hepatic injury and oxidative stress markers.a
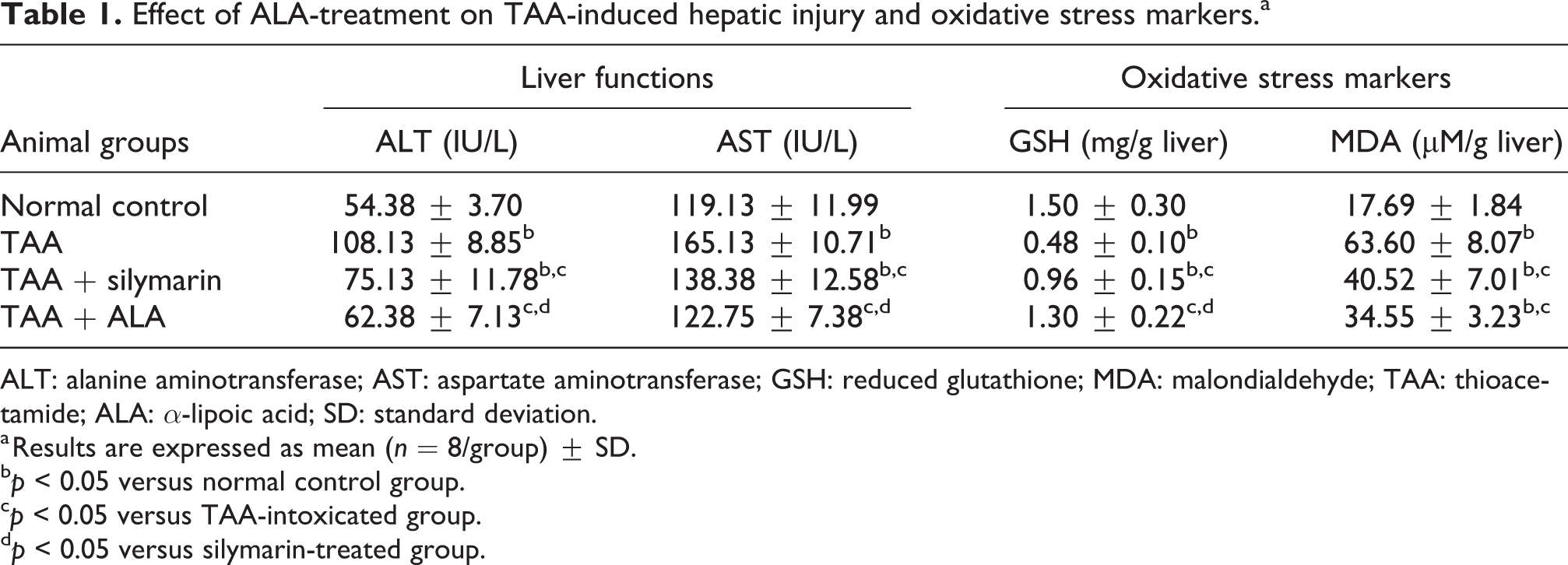
ALT: alanine aminotransferase; AST: aspartate aminotransferase; GSH: reduced glutathione; MDA: malondialdehyde; TAA: thioacetamide; ALA: α-lipoic acid; SD: standard deviation.
a Results are expressed as mean (n = 8/group) ± SD.
b p < 0.05 versus normal control group.
c p < 0.05 versus TAA-intoxicated group.
d p < 0.05 versus silymarin-treated group.
We further observed the effect of ALA on TAA-intoxicated liver sections stained with H&E (Figure 1(b) and (d)). Results revealed that TAA intoxication for 12 weeks caused distorted hepatic architecture with severe damage characterized by centrilobular necrosis and showing an elevated fibrosis score of almost stage 4 (S4, 3.67 ± 0.21). Meanwhile, treatment with silymarin partially recovered the damaged hepatic tissues, as denoted by reduced levels of necrosis and mitigated fibrosis scores of approximately S2, 1.83 ± 0.17. Notably, ALA treatment restored the normal hepatic architecture as confirmed by reduced fibrosis scores (approximately S1, 0.67 ± 0.21).
ALA suppressed TAA-induced oxidative stress in rat livers
TAA intoxication evoked oxidative stress, where increased MDA levels (259.53%) and depleted GSH contents (68.00%) were recorded in comparison to normal control rats. Administration of either silymarin or ALA noticeably reduced the elevated MDA level by 36.29% and 45.68%, respectively, associated with elevation of hepatic GSH contents by 100.00% and 170.83%, respectively, when compared to their corresponding TAA-intoxicated groups. Interestingly, ALA administration restored the hepatic GSH contents accompanied by an increase in its content by 35.42% in comparison with silymarin-treated group, as represented in Table 1.
ALA mitigated TAA-induced collagen deposition
Collagen deposition was determined through measuring the hepatic levels of TIMP-1 and HP (Figure 2(c) and (d)). Chronic TAA intoxication revealed a pronounced escalation in hepatic levels of TIMP-1 and HP by 2.42- and 4.24-fold, respectively, when compared to their corresponding normal control groups. Silymarin and ALA modulated the hepatic levels of both TIMP-1 (14.76% and 48.33%, respectively) and HP (47.96% and 55.84%, respectively) when compared to their corresponding TAA-intoxicated groups. In addition, ALA showed a considerable decline in TIMP-1 and HP hepatic levels by 39.40% and 15.16%, respectively, when compared to silymarin-treated group.

Effect of daily treatment of ALA on hepatic levels of (a) PDGF-BB and (b) TGF-β1, and (c) TIMP-1 and (d) HP levels in liver tissue homogenates. Data are expressed as mean (n = 6) ± SD; *p < 0.05 versus normal control group; † p < 0.05 versus TAA-intoxicated group; # p < 0.05 versus silymarin-treated group. TAA: thioacetamide; PDGF-BB: platelet-derived growth factor BB; TGF-β1: transforming growth factor-β1; TIMP-1: tissue inhibitor of matrix matalloproteinases type-1; HP: hydroxyproline; TAA: thioacetamide; ALA: α-lipoic acid; SD: standard deviation.
The diminished effect of ALA on TAA-induced collagen deposition was further confirmed via Sirius red staining of liver tissues (Figure 1(c)). TAA intoxication augmented collagen deposition as manifested by large fibrous septa forming micro- and macronodules around hepatic lobules. Administration of silymarin reduced the massive increase in collagen deposition that appeared in the form of mild, thin and moderate fibrous collagen bands, while ALA administration showed mild and thin fibrous bands.
ALA attenuated TAA-induced proliferation and activation of HSCs
Figure 2(a) and (b) illustrates the impact of ALA administration on hepatic levels of the profibrogenic cytokines PDGF-BB and TGF-β1 in TAA-intoxicated rats. Chronic TAA intoxication markedly elevated the hepatic levels of PDGF-BB and TGF-β1 by 171.43% and 197.65% when compared to their corresponding normal control groups. Treatment with either silymarin or ALA showed a marked decline in the hepatic levels of both PDGF-BB (31.57% and 49.12%; Figure 2(a)) and TGF-β1 (40.81% and 59.19%; Figure 2(b)), respectively, when compared to their corresponding TAA-intoxicated groups. ALA administration restored the normal hepatic TGF-β1 levels and resulted in a significant reduction in both PDGF-BB and TGF-β1 hepatic levels by 25.64% and 31.05% when compared to silymarin-treated group.
Next, HSCs activation was further investigated via immunohistochemical (IHC) staining of hepatic tissues with α-SMA antibody where the number of positively stained activated cells was quantified and represented in Figure 3(a) and (c). It was observed that α-SMA positively stained area was markedly increased (by 26.53-fold) after chronic TAA intoxication. Meanwhile, treatment with either silymarin or ALA significantly reduced the number of positively activated cells in comparison with TAA-injected rats by 75.47% and 83.78%, respectively. Furthermore, ALA resulted in a significant decline in the number of positively activated cells (by 33.87%) when compared to silymarin-treated rats.

Effect of ALA on α-SMA, caspase-3 and, LC3-II expressions in HSCs. Photomicrograph of IHC examinations of (a) α-SMA (×200) and (b) caspase-3 (×400) expressions in liver sections, quantitative estimation of positively stained cells with (c) α-SMA and (d) caspase-3, and quantitative estimation of mRNA expression of (e) LC3-II using qRT-PCR. Data are expressed as mean (n = 6) ± SD; *p < 0.05 versus normal control group; † p < 0.05 versus TAA-intoxicated group; # p < 0.05 versus silymarin-treated group. α-SMA: alpha-smooth muscle actin; TAA: thioacetamide; ALA: α-lipoic acid; SD: standard deviation; qRT-PCR: quantitative real-time polymerase chain reaction; IHC: immunohistochemical.
ALA induced apoptosis and suppressed autophagy in HSCs
HSCs apoptosis was investigated by means of caspase-3 IHC expression in TAA-intoxicated and TAA-treated hepatic tissues. Caspase-3 is a crucial mediator of apoptosis, known as executioner caspase, where a few numbers of caspase-3 positive cells (apoptotic cells) were detected in the normal control group and were significantly elevated in TAA-intoxicated group (87.5%). Caspase-3 positive cells were more enhanced in silymarin and ALA-treated groups by 100.00% and 172.88%, respectively, as compared to TAA-intoxicated hepatic sections. Furthermore, ALA resulted in a considerable elevation in caspase-3 expression by 36.44% in comparison to silymarin-treated group. The caspase-3 positively stained cells were mostly detected in the portal and periportal areas (areas of activated HSCs), where collagen deposition was identified (Figure 3(b) and (d)).
To examine the effect of ALA on the regulation of autophagy in TAA-induced liver fibrosis, the mRNA expression of LC3-II was detected in liver tissues using qPCR. Figure 3(e) indicates that the mRNA expression was extensively enhanced in TAA-intoxicated group by 762.92% when compared to the normal control group. Administration of either silymarin or ALA notably suppressed the expression of LC3-II by 37.24% and 44.01% when compared to the TAA-intoxicated group. Moreover, ALA showed further decline in its expressed levels by 10.79% when compared to silymarin (Figure 3(e)).
Discussion
ALA was reported to exert protective effects against chronic liver diseases through possessing powerful antioxidant and anti-inflammatory activities. 31 Although this is not the first study investigating the antifibrotic effects of ALA, however, its underlying antifibrotic mechanisms and its association with autophagy are elucidated herein. In addition, the investigated antifibrotic potential of ALA was compared to the reference drug silymarin. Silymarin is commonly used for the treatment of liver diseases both clinically 32 and in experimental animals. 33
Results of the present study confirmed that the administration of ALA daily for 8 weeks significantly attenuated chronic TAA-induced liver injury as indicated by restoration of serum levels of ALT and AST back to normal levels. These results run inconsistency with the recorded histopathological examinations herein where the hepatic sections of TAA-intoxicated rats showed a distorted hepatic architecture that appeared in the form of micro and macronodules, with an elevated fibrosis score of S4. Meanwhile, ALA restored the intact hepatic architectural pattern denoting the property of ALA in maintaining the integrity of the cell membrane of liver cells.
During chronic liver injury, Kupffer cells and hepatocytes release reactive oxygen species that increase the oxidative stress in hepatocytes, triggering their apoptosis and leading to the activation of HSCs. 34 In the current study, chronic TAA intoxication for 12 weeks was shown to be mediated by the liberation of free radicals as exemplified by a significant depletion in hepatic GSH stores associated with an increase in hepatic MDA levels reflecting the increase in lipid peroxidation as previously documented. 24 Administration of ALA modulated TAA-induced oxidative stress via replenishment of the hepatic GSH stores along with the marked reduction in hepatic MDA levels as formerly reported. 22
In this study, TAA-induced oxidative stress further triggered the activation and proliferation of HSCs via releasing TGF-β1 and PDGF-BB, the most potent profibrogenic cytokines, which in turn lead to the amplification of α-SMA expression. The obtained data indicate the progression of acute liver injury toward liver fibrosis as previously mentioned. 35 ALA ameliorated the upregulated hepatic levels of TGF-β1 and PDGF-BB, which resulted from TAA intoxication. These effects were accompanied by a noticeable decline in the number of activated HSCs as indicated by reduced expression of α-SMA. These findings indicate that ALA mitigates hepatic fibrosis at least partly via blocking the activation and proliferation of HSCs.
The pro-inflammatory cytokine TGF-β1 was evidenced to play an important role in the regulation of the production and deposition of ECM, mainly collagen type I, via inducing the activation of HSCs. 36 The activated HSCs consecutively synthesize TIMP-1, which regulates the alteration in hepatic ECM through inhibition of matrix metalloproteases. 37 TIMP-1 was also reported to correlate directly with the stage of hepatic fibrosis. 38 Results of the present study revealed that collagen deposition in TAA-induced liver fibrosis was attenuated post ALA treatment as illustrated by mild and thin fibrous collagen bands associated with reduction in hepatic HP levels, the major component of collagen. 39 These outcomes were further strengthened by considerable downregulation of hepatic levels of TIMP-1 in ALA-treated rats. Our results signify that ALA treatment attenuated liver fibrosis partly by remodeling the imbalance in collagen deposition that correlated with the recorded reduction in TGF-β1 levels.
Furthermore, autophagy was reported to participate in the progression of chronic liver disorders, 40 where TGF-β1 was reported to be a potent contributor in autophagy activation, thereby regulating HSCs proliferation and apoptosis. 41 In the present study, TAA intoxication stimulated autophagy of HSCs as indicated by elevated mRNA expression of LC3-II. Such increment in autophagy expression is in harmony with Hernandez-Gea et al. 9 who indicated that activation of HSCs triggers autophagy through providing the required energy resources for their activation, thus leading to the progression of liver fibrogenesis. Furthermore, we showed that the administration of ALA for 8 weeks counteracted this observed upregulated LC3-II expressed levels, which is in coherence with other studies that documented the blockade of autophagy in various cells after treatment with ALA. 21,42,43 Our data postulate that ALA modulates the progression of hepatic fibrosis via a possible correlation between autophagy suppression and inactivation of HSCs. Conversely, other studies showed that autophagic flux was enhanced during attenuation of hepatic fibrosis 44,45 through induction of HSC senescence. 46
Previously, it was shown that selective clearance of activated HSCs from the injured liver through induction of apoptosis contributes to the regression of hepatic fibrosis 47 Our results support this finding, where ALA treatment significantly triggered apoptosis of HSCs as denoted by increased caspase-3 expression in comparison to TAA-intoxicated rats. This could be explained in view of the relationship that exists between autophagy and apoptosis, 48 where the inhibition of autophagy precedes the induction of HSCs apoptosis, 49 which supports our findings reported herein. Furthermore, induction of apoptosis coincided with the recorded downstream in TIMP-1 levels and TGF-β1 blockade, thereby rendering activated HSCs more responsive to apoptosis. In concordance with our results, Troeger et al. 50 indicated that the increase in TIMP-1 expression was associated with inability of activated HSCs to undergo apoptosis resulting in downregulation of caspases.
Distinctly, in this study, ALA exhibited excelled activities over silymarin as exemplified by the improvement of liver functions, prominent antioxidant effects via restoration of the GSH stores, significant suppression of HSCs proliferation, activation, and autophagy. ALA as well showed distinguished decline in collagen deposition and enhanced the induction of HSCs apoptosis.
In conclusion, our data clearly show that the treatment of TAA-intoxicated rats with ALA did not only induce apoptotic clearance of activated HSCs but also halted the activation, proliferation, and autophagy of HSCs as well as declined collagen deposition within hepatic tissues. These findings provide mechanistic insights into the possible applicability of ALA as a new therapeutic approach for slowing down or reversing the progression of hepatic fibrosis, given that no effective antifibrotic therapies are currently available.
Footnotes
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This study is a part of a research project (ID-MS-99/A), which was financially supported by a grant from Theodor Bilharz Research Institute (Principal Investigator: NME-L).