
Abstract
Introduction
Liver fibrosis, occurring as a wound-healing response to maintain organ integrity, is generally resulted from the excessive accumulation of extracellular matrix (ECM). 1 It is also a pre-stage of liver cirrhosis, which is commonly caused by hepatitis virus infection, fatty liver disease, autoimmune hepatitis, schistosomiasis, and alcoholism.2,3 The continued progression of liver fibrosis causes distortion of the hepatic architecture and portal hypertension, which will eventually lead to cirrhosis, organ failure, and even hepatocellular carcinoma (HCC). 4 Cirrhosis and HCC are currently among the top 10 causes of death worldwide and lack effective treatment.5,6 Liver transplantation is now regarded as the only treatment method for end-stage cirrhosis, but the availability of donor organs is a major limitation.7,8
Hepatic stellate cell (HSC), a type of pleiotropic non-parenchymal cell that resides in the perisinusoidal space between hepatocytes and sinusoidal endothelial cells, plays a crucial role in liver fibrotic response. 9 In normal liver, HSCs show a quiescent phenotype storing vitamin A-rich lipid droplets. In response to liver injury, signals secreted by damaged hepatocytes and immune cells promote HSC activation and subsequent differentiation into myofibroblasts. Activated HSCs (aHSCs) are a major source of hepatic collagen and can remodel liver architecture by secreting ECM proteins, tissue inhibitors of metalloproteinases, and matrix metalloproteinases (MMPs).10,11 In chronic liver injury, prolonged and repeated activation of HSCs causes scar formation and perturbation of liver function and architecture, eventually leading to liver fibrosis.9,10 Thus, aHSC depletion is a major focus of the resolution of fibrosis. However, the underlying mechanisms of HSC activation in the pathological situation remain obscure.
Nicotinamide adenine dinucleotide (NAD+) is an essential co-enzyme in cellular redox reactions and also the substrate for multiple enzymes, including sirtuins and poly (ADP-ribose) polymerases (PARPs).12,13 NAD+ can be generated either through de novo pathway from tryptophan or salvage pathway from three NAD+ precursors, nicotinamide (NAM), nicotinic acid (NA), and nicotinamide riboside (NR). 14 However, in mammalian cells, the majority of NAD+ is produced from NAM through the salvage pathway. NAM is first converted to nicotinamide mononucleotide (NMN) in the rate-limiting step catalyzed by nicotinamide phosphoribosyltransferase (NAMPT) and then further converted into NAD+ by nicotinamide mononucleotide adenylyltransferases (NMNAT1-3).15,16 NAMPT has been implicated in regulating various cellular processes (i.e., oxidative stress response, lipid and glucose metabolism and inflammation) involved in the pathogenesis of metabolic disorders through influencing the activity of NAD+-dependent enzymes, such as sirtuins and PARPs.17,18 Moreover, accumulating evidence has shown that NAMPT plays a critical role in modulating the development of diet- and alcohol-induced hepatic steatosis.19–21 Interestingly, a recent study has demonstrated lower NAMPT protein abundance in CCl4-induced fibrotic mouse liver samples, indicating NAMPT might be an important regulator for progression of liver fibrosis. 22 However, the effects of NAMPT on the pathogenesis of liver fibrosis, especially on HSC activation are largely unknown. In this study, we aimed to determine the role of NAMPT-mediated NAD+ salvage pathway in regulating HSC activation and the development of CCl4-induced liver fibrosis in mice.
Materials and methods
Animal studies
The study was approved by the Institutional Animal Care and Use Committee of Xinxiang Medical University, China. All animal procedures were performed in accordance with “Guide for the Care and Use of Laboratory Animals” published by the National Institutes of Health. Euthanasia was performed using compressed carbon dioxide (CO2) from gas cylinders or cervical dislocation under anesthesia.
C57BL/6N mice were purchased from Charles River (Beijing, China). Mice were maintained in an environmentally controlled animal facility and fed a rodent chow with ad libitum access to water. To induce liver fibrosis, 6–8-week-old male mice were intraperitoneally injected with a 10% solution of CCl4 in olive oil (5 μL/g BW) twice a week for 6 weeks. Control mice were intraperitoneally injected with the same volume of olive oil at the same time intervals. For adenovirus-mediated gene delivery, adenoviruses carrying NAMPT, green fluorescent protein (GFP) were injected into mice at a dose of 1 × 10 9 pfu via tail vein. For NMN treatment, mice were intraperitoneally injected with NMN (200 mg/kg/day) every day for 2 weeks.
Realtime RT-PCR
Primer sequences used in RT-PCR.

Cell culture and primary HSC isolation
The LX2 cells were a generous gift from Dr. Yong Liao of Chongqing Medical University. LX2 cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM, 4.5 g/L glucose), 10% fetal bovine serum (FBS), and penicillin/streptomycin. For transforming growth factor-β1 (Tgfβ1) treatment, LX2 cells were treated with 10 ng/mL Tgfβ1 for 48 h. HSCs were isolated from mice as described previously. 23 Briefly, mouse liver was sequentially perfused with ethylene glycol tetraacetic acid (EGTA), pronase (Sigma Aldrich) and collagenase (Roche) solutions. Then, the liver was minced, transferred to pronase/collagenase solution containing 1% (vol/vol) DNase I (Roche), and shaken for 30 min at 37°C. Next, HSCs were separated by Nycodenz (Axis-Shield) density gradient and plated in 6-well dishes and then cultured in DMEM medium with 10% FBS.
Western blot analysis
Protein extracts from cells or liver tissues were made in radio-immunoprecipitation assay (RIPA) lysis buffer (Beyotime, China). Protein extracts were resolved on a sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) gel and transferred to nitrocellulose membranes. The membranes were blocked in Tris buffered saline with Tween-20 (TBST) containing 5% skimmed milk for 1 h at RT and incubated overnight at 4°C with the following primary antibodies: anti-NAMPT, anti-actinin, and anti-α-smooth muscle actin (anti-αSMA, Proteintech). The membranes were washed 3 times with TBST before incubation with horseradish peroxidase (HRP)–conjugated secondary antibody at RT for 1 h. After three washes in TBST, the immune complexes were detected using the electrochemiluminescence (ECL) detection reagents (Sigma).
NAD+ measurement
NAD+ levels in cells and liver samples were measured with an enzymatic cycling assay as previously described. 21 Briefly, cells (1 × 106/reaction) or liver tissues (50 mg/reaction) were lysed in acid extraction buffer (50 mM HCl, 1 mM EDTA, 300 μL for each NAD+ assay). The samples were incubated at 60°C for 10 min to destroy endogenous enzyme activities and pyridine nucleotides and neutralized by 0.4 M Tris base (75μL for each sample). Then, the samples were centrifuged at 12,000 g for 5 min at 4°C and the supernatants were collected. Next, 5 μL of each supernatant was mixed with 85 μL cycling buffer (10 mM Tris, pH 8.0, 5 mM EDTA, 0.5 mM MTT, 0.2 mg/mL alcohol dehydrogenase (ADH, Sigma Aldrich), and 1 mM PES). The cycling reaction was initiated by adding 10 μL 6 M ethanol to each assay, and the absorbance at 570 nm was measured using a plate reader (Molecular Devices).
Sirius red staining
Mouse liver tissues were dissected, rapidly fixed in 4% paraformaldehyde, and then routinely processed for paraffin embedding, and 5 μm sections were cut and mounted on glass slides. Deparaffinized sections were stained with picro-sirius red stain solution (0.1% direct red 80 plus 0.1% fast green FCF dissolved in saturated aqueous picric acid) for 1 h at RT. The sections were dehydrated, cleared in xylene, and mounted with a coverslip. Images were captured using a Leica DM1000 microscope equipped with an EC3 digital camera.
Analysis of serum ALT levels
Serum alanine aminotransferase (ALT) levels were determined using assay kit from Nanjing Jiancheng Bioengineering Institute according to the manufacturer’s instruction. Briefly, 10 μL of serum was incubated with a 200 μL mixture of reagent A (160μL) and B (40μL) provided by the kit. Absorbance at 340 nm was immediately recorded at 1 min intervals for 5 min. The slope of absorbance decrease is proportional to ALT activity.
Adenovirus preparation
Adenoviral constructs expressing NAMPT or GFP were generated using the pAdEasy system (Agilent); adenoviruses were amplified in HEK293A cells and purified by cesium chloride (CsCl) gradient centrifugation. The purified adenoviruses were titered using an Adeno-XTM Rapid Titer kit (Takara) according to the manufacturer’s manual. In general, we used 25–50 multiplicity of infection (moi) for overexpression in LX2 cells and in primary HSCs.
Statistical analysis
All data are expressed as the mean ± SEM. Analysis was performed using 2-tailed unpaired Student’s t test, and a value of p < .05 was considered as significant.
Results
Nampt expression is decreased in CCl4-induced fibrotic liver
Mice receiving CCl4 treatment for 6 weeks developed severe fibrosis as shown in a representative Sirius red stained image (Figure 1(a)). Interestingly, CCl4 treatment led to a significant reduction of hepatic NAD+ levels as compared to oil-treated controls (Figure 1(b)). As expected, after CCl4 treatment, increased αSMA expression was detected, indicating forming fibrous tissue while, in contrast, Nampt mRNA and protein levels were remarkably reduced (Figure 1(c) and (d)). NAMPT and NAMPT-mediated NAD+ biosynthesis is compromised in CCl4-induced fibrotic mouse livers. C57BL/6N male mice (6–8-week-old) were intraperitoneally injected with a 10% solution of CCl4 in olive oil (5 μL/g BW) or vehicle (olive oil) twice a week for 6 weeks (n = 4–6 mice for each group). (a) Paraffin-embedded liver sections from oil or CCl4-treated mice were stained with Sirius red. (b) NAD+ levels were measured in liver samples. (c) Western blot analysis of NAMPT and αSMA expression in liver samples. (d) qPCR analysis of Nampt expression in liver samples. Data are shown as mean ± SEM *p< .05. Scale bars: 50 μm.
Tgfβ1 downregulates NAMPT expression in LX2 cells
The LX2 human hepatic stellate cell line exhibits key features typical of stellate cells, making them a highly suitable model of human hepatic fibrosis.
24
Tgfβ1, a key stimulator for HSC activation, remarkably induced expression of profibrogenic markers, such as ACTA2, COL1A1, and TIMP1 (Figure 2(a) and (c)). In contrast, Tgfβ1 treatment significantly reduced Nampt expression (Figure 2(a) and (b)) as well as intracellular NAD+ levels in LX2 cells (Figure 2(d)). Tgfβ1 decreases NAMPT-mediated NAD+ biosynthesis and the expression of profibrogenic genes in LX2 cells. LX2 cells were cultured in DMEM (4.5 g/L glucose) with 10% FBS and treated with 10 ng/mL Tgfβ1 for 48 h (n = 3 for each group). (a) Western blot analysis of NAMPT and αSMA expression in LX2 cells treated with vehicle or Tgfβ1. (b) qPCR analysis of Nampt expression in LX2 cells. (c) Expression of profibrogenic genes was analyzed by qPCR in LX2 cells. (d) Intracellular NAD+ levels were analyzed in LX2 cells. Data are shown as mean ± SEM *p< .05.
NAMPT overexpression alleviates CCl4-induced liver fibrosis in mice
To determine whether increased NAMPT expression can protect against CCl4-induced liver fibrosis, NAMPT-expressing (Ad-NAMPT) or control GFP (Ad-GFP) adenoviruses were injected into the tail vein of the mice that had been treated with CCl4 or olive oil (control) for 4 weeks. After adenovirus administration, mice were kept on treating with CCl4 or oil for another 2 weeks and then were sacrificed for blood and tissue collection. Adenovirus-transduced NAMPT-HA expression in liver was around 5 times higher than endogenous Nampt expression (Figure 3(a)). Accordingly, the hepatic NAD+ levels were upregulated by ∼34% and ∼58% in NAMPT overexpressed oil-treated and CCl4-treated mouse livers, respectively (Figure 3(b)). Serum ALT levels were dramatically increased by CCl4 treatment, whereas NAMPT overexpression significantly lowered ALT levels in CCl4-treated group but not in oil-treated control group (Figure 3(e)). Indeed, NAMPT overexpression in the liver also reduced collagen deposition (Figure 3(c) and (d)) as well as expression of profibrogenic genes in CCl4-treated mice (Figure 3(f)). Adenovirus-mediated NAMPT overexpression alleviates CCl4-induced liver fibrosis in mice. Ad-NAMPT or control Ad-GFP adenoviruses were injected into the tail vein of mice that had been treated with CCl4 or olive oil for 4 weeks. Mice were kept on treating with CCl4 or oil for another 2 weeks and then were sacrificed for blood and tissue collection (n = 5 mice for each group). (a) Western blot analysis of NAMPT and αSMA expression in liver samples from Ad-NAMPT or Ad-GFP administrated mice. (b) NAD+ levels were examined in mouse liver tissues. (c) Sirius red staining of paraffin-embedded liver sections. (d) Sirius red positive staining was quantified using ImageJ software. (e) Serum ALT levels were analyzed. (f) Expression of profibrogenic genes was analyzed by qPCR in liver samples. Data are shown as mean ± SEM *p< .05. Scale bars: 50 μm.
NAMPT overexpression in HSCs suppresses the expression of profibrogenic genes
As we know, adenovirus vectors are mainly sequestered by the liver after intravenous administration.
25
Theoretically, any cells in liver, including parenchymal and non-parenchymal cells, that possess adenovirus receptor can be infected by adenovirus. To verify whether HSCs had been infected by adenovirus after intravenous administration, we isolated primary HSCs from Ad-NAMPT or Ad-GFP adenoviruses administered mice. After isolation, the primary HSCs were kept in culture for 6 days. During this period, HSCs became activated and transdifferentiated into myofibroblast-like cells. Then, cells were collected for further analysis. As shown in Figure 4(a), adenovirus-mediated NAMPT-HA expression in primary HSCs was about 2 times higher than endogenous Nampt expression. Importantly, NAMPT overexpression in primary HSCs significantly elevated intracellular NAD+ content, whereas decreased the expression of profibrogenic markers (Figure 4(b) and (c)). Thus, adenovirus-mediated NAMPT overexpression in HSCs contributed to the hepatic anti-fibrotic effects of intravenous administration (tail vein) of Ad-NAMPT adenovirus in mice. To further demonstrate that NAMPT indeed plays an important role in HSC activation, we also overexpressed NAMPT in Tgfβ1 stimulated LX2 cells. Adenovirus-mediated NAMPT-HA expression in LX2 cells was about 4 times higher than endogenous Nampt expression (Figure 4(d)). As a result, intracellular NAD+ levels were increased by 45% in NAMPT overexpressed cells (Figure 4(e)). In line with what we observed in primary HSCs, NAMPT overexpression also remarkably decreased the expression of ACTA2, COL1A1, and TIMP1 (Figure 4(f)). NAMPT overexpression in HSCs increases NAD+ levels and suppresses the expression of profibrogenic genes. Mice were injected with Ad-NAMPT or Ad-GFP adenoviruses via tail vein. 3 days after injection, 2 Ad-NAMPT and 2 Ad-GFP administrated mice were used for HSC isolation. HSCs isolated from each group were pooled together and plated in 6-well dish and cultured in DMEM (4.5 g/L glucose) with 10% FBS for 6 days (n = 3 for each group). LX2 cells were also cultured in DMEM (4.5 g/L glucose) with 10% FBS and treated with 10 ng/mL Tgfβ1 for 48 h (n = 6 for each group). (a) Western blot analysis of NAMPT and αSMA expression in primary HSCs isolated from Ad-NAMPT or Ad-GFP administrated mice. (b) NAD+ levels were examined in primary HSCs. (c) Expression of profibrogenic genes (e.g. Acta2, Col1a1, and Timp1) was analyzed by qPCR in primary HSCs. (d) Western blot analysis of NAMPT and αSMA expression in LX2 cells infected with Ad-NAMPT or Ad-GFP. (e) NAD+ levels were examined in LX2 cells. (f) Expression of profibrogenic genes was analyzed by qPCR in LX2 cells. Data are shown as mean ± SEM *p< .05.
NMN treatment inhibits LX2 cell activation
As we shown in Figure 1(b) and Figure 2(d), NAMPT-mediated NAD+ biosynthesis is compromised in CCl4-induced fibrotic livers or in Tgfβ1-treated LX2 cells. NMN, a product of the NAMPT reaction in NAD+ salvage pathway, has been shown to ameliorate metabolic disorders including hepatic inflammation and steatosis by restoring NAD+ levels.
26
We then determined the effects of NMN on LX2 activation. Tgfβ1 stimulated LX2 cells were treated with vehicle (PBS) or NMN for 12 h. As expect, NMN treatment increased intracellular NAD+ levels in LX2 cells (Figure 5(a)). Importantly, NMN treatment remarkably decreased the expression of profibrogenic genes but has no effect on NAMPT expression (Figure 5(b) and (c)). NMN treatment reduces the expression of profibrogenic genes in LX2 cells. LX2 cells were treated with NMN (200 μM) for 12 h (n = 4–5 for each group). (a) NAD+ levels were examined in LX2 cells after NMN treatment. (b) Western blot analysis of NAMPT and αSMA expression in LX2 cells. (c) Expression of profibrogenic genes was analyzed by qPCR in LX2 cells. Data are shown as mean ± SEM *p< .05.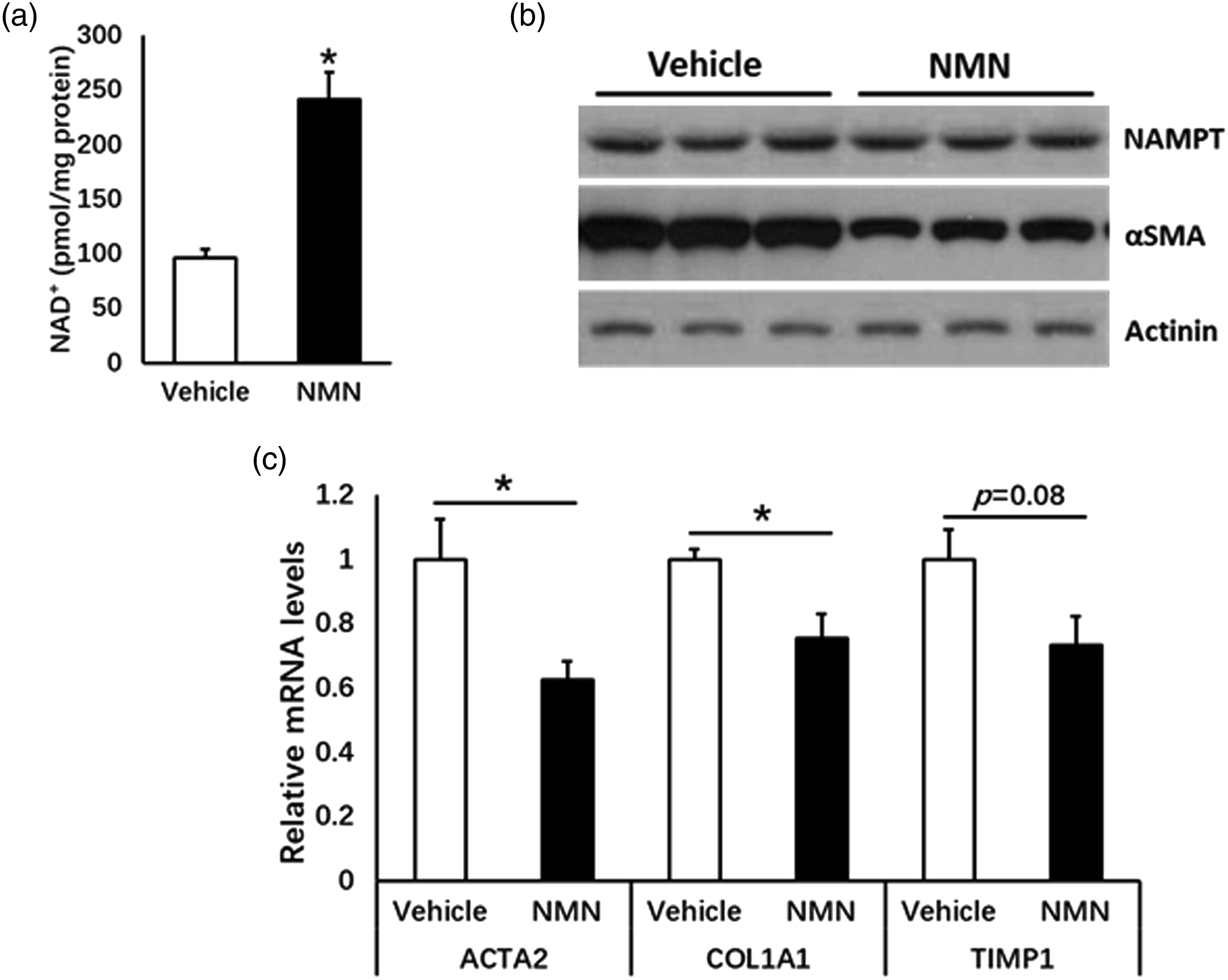
NMN administration protects against the development of CCl4-induced liver fibrosis
We next tested whether NMN also has protective effects on CCl4-induced mouse model of fibrosis. Mice which had been treated with CCl4 for 4 weeks were used for NMN treatment. Mice were administrated with NMN (200 mg/kg/day, IP injection) every day for 2 weeks. During this period, we continued to treat mice with CCl4 to keep developing fibrosis. After 2 weeks of NMN administration, mice were sacrificed for blood and tissue collection. As expected, NMN treatment dramatically elevated hepatic NAD+ content but did not affect Nampt expression (Figure 6(a) and (b)). Importantly, serum ALT levels and liver collagen deposition induced by CCl4 were significantly reduced after a 2-week NMN treatment (Figure 6(c), (d) and (e)). NMN administration protects against the development of CCl4-induced liver fibrosis. CCl4-treated mice were daily administrated with NMN via IP injection (200 mg/kg/day) (n = 4–5 mice for each group). (a) NAD+ levels were examined in liver tissues from mice treated with vehicle or NMN. (b) Western blot analysis of NAMPT and αSMA expression in liver samples. (c) Serum ALT levels were analyzed. (d) Sirius red staining of paraffin-embedded liver sections. (e) Sirius red positive staining was quantified using ImageJ software. Data are shown as mean ± SEM *p< .05. Scale bars: 50 μm.
Discussion
A decline in cellular NAD+ levels is tightly related to the development of various metabolic disorders, including diabetes and fatty liver disease.12–14 Importantly, compromised NAD+ biosynthesis mediated by NAMPT has been shown to contribute to the development of early stages of liver damage.19,20 Notably, our previous study has demonstrated hepatic overexpression of NAMPT can alleviate alcohol-induced liver steatosis and inflammation. 21 Intriguingly, accumulating evidence has shown that boosting NAD+ levels by supplementing NAD+ intermediates, such as NAM, NMN, and NR, can ameliorate age-associated pathological abnormalities and fatty liver disease.26–28 Mitchell et al. have revealed that supplementing NAM protects the liver function, glucose metabolism, and overall health of aging mice. 27 Moreover, Yoshino et al. have shown that systemic NMN administration effectively enhances NAD+ biosynthesis in various peripheral tissues (including liver) and treats the pathophysiology of diet- and age-induced liver steatosis and metabolic disorders. 26 In addition, NR supplementation has been found to protect against high fat diet–induced fatty liver and metabolic disorders in mice. 28
Upon stimulation by liver injury, quiescent HSCs are activated and believed to be the principle cell type responsible for producing ECM proteins during the progression of liver fibrosis.9,10 The HSCs are non-parenchymal cells that reside in the space of Disse in the liver and comprise a population of less than 8% of total liver cells.9,10 However, whether NAMPT catalyzed NAD+ salvage pathway contributes to the activation of HSCs and progression of liver fibrosis remains largely unknown. The results presented in our study showed that NAMPT-mediated NAD+ biosynthesis was compromised in the fibrotic mouse livers induced by CCl4 and in LX2 cells activated by Tgfβ1. We also demonstrated that promoting NAD+ biosynthesis by NAMPT overexpression or by NNM administration effectively ameliorated HSC activation and liver fibrosis in mice. Intriguingly, two studies concerning the role of NR on liver fibrosis have been published recently.29,30 The study done by Jiang et al. reported that NR supplementation prevented liver fibrosis via suppressing activation of HSC, and this protective effect was mediated by reducing the acetylation of transcription factors Smad2 and Smad3. 29 Impressively, Pham et al. showed that NR inhibited HSC activation, leading to reduced liver fibrosis in a diet-induced mouse model of liver fibrosis. 30 Collectively, both our and their studies have confirmed the notion that augmentation of NAD+ levels in HSCs by either overexpression of NAMPT or by administration with NAD+ precursors (NMN or NR) is a good method to suppress HSC activation and thereby ameliorate liver fibrosis.
Activation of HSC is believed to be a key step in the development of liver fibrosis. 9 Through its NAD+-biosynthesis activity, NAMPT modulates the activity of NAD+-dependent enzymes, such as sirtuin family, and thereby regulates various cellular processes. 17 The downstream targets mediate the effects of NAMPT overexpression on HSC activation still need to be elucidated. Among the seven sirtuin family members in mammals, SIRT1 is the best characterized regulator in metabolic processes. 31 Several studies have shown that SIRT1 might play critical roles in downregulating HSC activation and antagonizing liver fibrosis via various pathways.32–34 In addition to SIRT1, there might be some other NAD+-sensitive or even NAD+ consuming factors that also contribute to the effects of NAD+ restoration in HSC. Notably, SIRT2 inhibition results in the degradation of c-MYC, thereby suppressing HSC activation. 35
Apoptosis of damaged hepatocytes stimulates HSC activation either directly by phagocytosis of apoptotic bodies or indirectly by damage-related molecular patterns. 36 Adenovirus-mediated gene delivery predominantly targets to liver, including hepatocytes and other non-parenchymal cells, for example, HSCs. 25 Thus, intravenous administration of Ad-NAMPT resulted in adenovirus-mediated NAMPT expression not only in HSCs, but also in hepatocytes. As we have shown, NAMPT overexpression in HSCs upregulated NAD+ levels and inhibited the expression of profibrogenic genes, which contributed to the alleviation of liver fibrosis. Importantly, a study has shown that NAMPT exerts anti-apoptotic effects in hepatocytes. 37 Thus, adenovirus-mediated NAMPT overexpression in hepatocytes may also contribute to the ameliorated liver fibrosis through inhibiting hepatocyte apoptosis induced by CCl4 treatment. Therefore, in our study, the amelioration of CCl4-induced liver fibrosis should be attributed to the augmentation of NAD+ levels not only in HSCs but also in hepatocytes (and/or other cell types) via NAMPT overexpression or NMN administration.
In summary, the present study showed that NAMPT-mediated NAD+ biosynthesis is compromised in activated HSCs and in CCl4-induced fibrotic livers. Overexpression of NAMPT or administration with NMN restores NAD+ levels, alleviates the activation of HSCs, and thereby protects against CCl4-induced liver fibrosis. Taken together, our results indicate that activation of NAMPT or administration of NMN might be a potential intervention against liver fibrosis.
Footnotes
Author contributions
LX and CY performed the majority of the experiments, analyzed data, and wrote the article. JM performed histological analysis and photography of the samples. XZ assited with animal breeding and genotyping. QW designed the experiments, analyzed, and interpreted the data and revised the article. XX designed and performed the experiments, analyzed data, and wrote the article.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by grants from Program for Science & Technology Innovation Talents in Higher Education of Henan Province (20HASTIT046), National Natural and Science Foundation of China (U1904132), Key Scientific Research Project of Universities in Henan (19A180005), Key Science and Technology Project of Xinxiang (GG2019008), Training Program for Excellent Young Teachers in Higher Education of Henan province (2017GGJS110).