
Abstract
Amitriptyline is a widely used tricyclic antidepressant, which acts primarily as a serotonin–norepinephrine reuptake inhibitor. This study examined the effect of amitriptyline on Ca2+ homeostasis and its related mechanism in MG63 human osteosarcoma cells. Amitriptyline evoked cytosolic-free Ca2+ concentrations ([Ca2+]i) rises concentration dependently. Amitriptyline-evoked Ca2+ entry was confirmed by Mn2+-induced quench of fura-2 fluorescence. This entry was inhibited by Ca2+ entry modulators nifedipine, econazole, SKF96365, the protein kinase C (PKC) activator phorbol 12-myristate 13 acetate but was not affected by the PKC inhibitor GF109203X. In Ca2+-free medium, treatment with the endoplasmic reticulum Ca2+ pump inhibitor thapsigargin (TG) inhibited amitriptyline-evoked [Ca2+]i rises by 95%. Conversely, treatment with amitriptyline abolished TG-evoked [Ca2+]i rises. Inhibition of phospholipase C (PLC) with U73122 inhibited amitriptyline-evoked [Ca2+]i rises by 70%. Amitriptyline killed cells at 200–500 μM in a concentration-dependent fashion. Chelating cytosolic Ca2+ with 1,2-bis(2-aminophenoxy)ethane-N, N, N′, N′-tetraacetic acid/AM did not reverse amitriptyline-induced cytotoxicity. Collectively, our data suggest that in MG63 cells, amitriptyline induced [Ca2+]i rises by evoking PLC-dependent Ca2+ release from the endoplasmic reticulum and Ca2+ entry via PKC-regulated store-operated Ca2+ entry. Amitriptyline also induced Ca2+-disassociated cell death.
Introduction
Amitriptyline is one of the first “reference” tricyclic antidepressants. 1 Over the past 40 years, a number of newer tricyclics, heterocyclics, and selective serotonin reuptake inhibitors have been introduced. 2 Amitriptyline acts primarily as a serotonin–norepinephrine reuptake inhibitor, with strong actions on the serotonin transporter and moderate effects on the norepinephrine transporter. 1,2 It has negligible influence on the dopamine transporter and therefore does not affect dopamine reuptake, being nearly 1000 times weaker on it than on serotonin. 1,2 In vitro, diverse physiological effects have been reported for amitriptyline. Amitriptyline was shown to alter smooth muscle reactivity in isolated rat trachea, 3 inhibit D-cyclin transactivation, 4 and induce myeloma apoptosis by inhibiting histone deacetylases in human myeloma cells. 4 In Ca2+ signaling, amitriptyline was shown to affect Ca2+ homeostasis in different models. Previous studies have shown that amitriptyline inhibited voltage-dependent Ca2+ channels and Na+-Ca2+ exchange in rat brain cortex synaptosomes, 5 reduced L-type Ca2+ channel current in mouse trigeminal ganglion neurons, 6 and altered sarcoplasmic reticulum Ca2+ handling in ventricular myocytes. 7,8 Besides inhibitory effect of amitriptyline, it has been shown that amitriptyline increased cytosolic-free Ca2+ concentrations ([Ca2+]i) in glioma C6 cells 9 and Chinese hamster ovary (CHO) cells transfected with the human 5-HT2C receptors. 10 However, the effect of amitriptyline on Ca2+ homeostasis and its physiology in most cells including human osteosarcoma cells is unclear.
Ca2+ ions play a pivotal role in various biological responses. Rises in [Ca2+]i can trigger many pathophysiological cellular responses. Therefore, if a chemical induces a Ca2+ signal in any cell type, it necessitates the effort to examine the underlying pathways because this chemical may cause significant physiological changes in this cell. 11,12 Inositol 1,4,5-trisphosphate (IP3), derived from activation of phospholipase C (PLC), is a predominant second messenger for releasing store Ca2+ from the endoplasmic reticulum. 11,12 Movement of store Ca2+ may stimulate Ca2+ influx across the plasma membrane via store-operated Ca2+ entry. 11,12 On the other hand, uncontrolled [Ca2+]i rises induce malfunction of enzymes, genes, secretion, contraction, apoptosis, proliferation, and so on. 11,12 In order to explore the effect of amitriptyline on [Ca2+]i in human osteosarcoma cells, the MG63 cell line was used because it induced significant [Ca2+]i elevation when stimulated by many chemicals. It has been reported that in this cell line, [Ca2+]i rises can be triggered in response to the addition of various chemicals such as NPC-14686, 13 sertraline, 14 and 3,3′-diindolylmethane. 15
Using fura-2 as a Ca2+-sensitive fluorescent probe, we observed that amitriptyline induced [Ca2+]i rises both in the presence and absence of extracellular Ca2+ in MG63 cells. The aim of this study was to evaluate the concentration–response relationships and explore the pathways underlying amitriptyline-induced Ca2+ entry and Ca2+ release. The effect of amitriptyline on cell viability was also examined.
Materials and methods
Chemicals
The chemicals for cell culture were purchased from Gibco® (Gaithersburg, Maryland, USA). Aminopolycarboxylic acid/acetoxy methyl (fura-2/AM) and 1,2-bis(2-aminophenoxy)ethane-N, N, N′, N′-tetraacetic acid/acetoxy methyl (BAPTA/AM) were from Molecular Probes® (Eugene, Oregon, USA). All other chemicals were from Sigma-Aldrich® (St. Louis, Missouri, USA) unless otherwise stated.
Cell culture
MG63 human osteosarcoma cells obtained from Bioresource Collection and Research Center (Taiwan) were cultured in Minimum Essential Medium (MEM) medium supplemented with 10% heat-inactivated fetal bovine serum, 100 U/ml penicillin and 100 μg/ml streptomycin.
Solutions applied in [Ca2+]i assays
Ca2+-containing medium (pH 7.4) had 140 mM NaCl, 5 mM KCl, 1 mM MgCl2, 2 mM CaCl2, 10 mM 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid, and 5 mM glucose. Ca2+-free medium had similar chemicals as Ca2+-containing medium except that CaCl2 was replaced with 0.3 mM ethylene glycol tetraacetic acid (EGTA) and 2 mM MgCl2. Amitriptyline was dissolved in ethanol as a 2 M stock solution. The other chemicals were dissolved in water, ethanol, or dimethyl sulfoxide. The concentration of organic solvents in the experimental solutions did not exceed 0.1% and did not influence viability or [Ca2+]i.
[Ca2+]i assays
The [Ca2+]i was measured as previously described. 13 –15 Confluent cells grown on 6 cm dishes were trypsinized and suspended in medium at a concentration of 106 cells per milliliter. Cell viability was assayed by trypan blue exclusion (adding 0.2% trypan blue to 0.1 ml cell suspension). The viability was found to be >95%. Cells were subsequently loaded with 2 μM fura-2/AM for 30 min at 25°C in the same medium. Afterwards, cells were washed with Ca2+-containing medium twice and suspended in Ca2+-containing medium at a concentration of 107 cells per milliliter. Fura-2 fluorescence measurements were performed in a water-jacketed cuvette (25°C) with continuous stirring; the cuvette contained 0.5 million cells suspended in 1 ml of medium. The Shimadzu RF-5301PC spectrofluorophotometer was used to determine the fluorescence after 0.1 ml cell suspension was added to 0.9 ml medium. The signal was recorded at excitation signals of 340 and 380 nm and emission signal of 510 nm at 1-s intervals. During the recording, Ca2+-associated modulators (nifedipine, econazole, SKF96365, phorbol 12-myristate 13 acetate (PMA), GF109203X, thapsigargin (TG), and U73122) were added to the cuvette by pausing the recording for 2 s to open and close the cuvette-containing chamber. After the experiments, Triton X-100 (0.1%) and CaCl2 (5 mM) were added to the cuvette to gain the maximal fura-2 fluorescence. The Ca2+ chelator EGTA (10 mM) was subsequently added to chelate Ca2+ in the cuvette to obtain the minimal fura-2 fluorescence. Pioneering experiments showed that cells bathed in a cuvette had a viability of 95% after 30 min of fluorescence recording. [Ca2+]i was calculated as previously reported. 16
Mn2+ quenching of fura-2 fluorescence was conducted in Ca2+-containing medium which contained 50 μM MnCl2. MnCl2 was added to cell suspension in the cuvette at time point of 30 s prior to the fluorescence recoding. Data were collected at excitation signal at 360 nm (Ca2+-insensitive) and emission signal at 510 nm at 1-s intervals as described previously. 17
Cell viability assays
Cell viability was determined depending on the ability of dehydrogenases in the cells to cleave tetrazolium salts. 13 –15 Elevations in the intensity of developed color were found to correlate with live cell number. Measurements were performed according to manufacturer’s manuals (Roche Molecular Biochemical, Indianapolis, Indiana, USA). Cells were seeded in 96-well plates at a concentration of 10,000 cells per well in medium for 24 h in the presence of amitriptyline. WST-1 (4-[3-[4-lodophenyl]-2-4(4-nitrophenyl)-2H-5-tetrazolio-1,3-benzene disulfonate]; 10 μl pure solution), a cell viability detecting reagent, was added to samples after amitriptyline treatment, and cells were incubated for 30 min in a humidified atmosphere. In experiments that required chelation of cytosolic Ca2+, cells were treated with 5 μM BAPTA/AM for 1 h prior to incubation with amitriptyline. The cells were washed once with medium and incubated in the presence or absence of amitriptyline for 24 h. An enzyme-linked immunosorbent assay reader was applied to determine the absorbance of samples (A450). Optical intensity was normalized and presented as a percentage of the control.
Statistics
Mean ± standard error mean of three separate experiments was used to present the data. Data were analyzed by one-way analysis of variances using the Statistical Analysis System (SAS®; SAS Institute Inc., Cary, North Carolina, USA). Multiple comparisons between group means were performed by post hoc analysis using Tukey’s honestly significantly difference procedure. A p value of <0.05 was considered significant.
Results
Effect of amitriptyline on [Ca2+]i
Figure 1(a) illustrates that the baseline [Ca2+]i level was 51 ± 1 nM. Amitriptyline induced [Ca2+]i rises in a concentration-dependent manner between 200 μM and 1000 μM in Ca2+-containing medium. Amitriptyline evoked [Ca2+]i rises that attained to a net increase of 81 ± 2 nM (n = 3) at a concentration of 1000 μM. The Ca2+ signal saturated at 1000 μM amitriptyline since 1500 μM amitriptyline did not evoke a larger response (not shown). Figure 1(b) depicts that in Ca2+-free medium, 200–1000 μM amitriptyline induced concentration-dependent [Ca2+]i rises. The concentration–response relationship of amitriptyline-induced [Ca2+]i rises was shown in Figure 1(c). The EC50 value was 400 ± 3 μM or 452 ± 2 μM in Ca2+-containing medium or Ca2+-free medium, respectively, by fitting to a Hill equation.

Effect of amitriptyline on [Ca2+]i in fura-2-loaded MG63 cells. (a) Amitriptyline was added at 25 s. The concentration of amitriptyline was indicated. The experiments were performed in Ca2+-containing medium. (b) Effect of amitriptyline on [Ca2+]i in the absence of extracellular Ca2+. Amitriptyline was added at 25 s in Ca2+-free medium. (c) Concentration–response plots of amitriptyline-induced [Ca2+]i rises in the presence or absence of extracellular Ca2+. y-axis is the percentage of the net (baseline subtracted) area under the curve (25–250 s) of the [Ca2+]i rises induced by 1000 μM amitriptyline in Ca2+-containing medium. Data are mean ± SEM of three experiments. *p < 0.05 compared to open circles; Ca2+: calcium ion; SEM: standard error mean.
Amitriptyline-induced Mn2+ influx
To exclude the possibility that the smaller amitriptyline-induced response in Ca2+-free medium was caused by 0.3 mM EGTA-induced depletion of intracellular Ca2+, experiments were conducted to confirm that Ca2+ influx participated in amitriptyline-evoked [Ca2+]i rises. Mn2+ enters cells through similar pathways as Ca2+ but quenches fura-2 fluorescence at all excitation wavelengths. 17 Thus, Ca2+ entry can be indirectly deduced by quenching fura-2 fluorescence excited at the Ca2+-insensitive excitation wavelength of 360 nm by Mn2+. In the following experiments, the effect induced by 1000 μM amitriptyline was used as control because amitriptyline-induced Ca2+ response saturated at 1000 μM. Figure 2 illustrates that 1000 μM amitriptyline triggered an immediate reduce in the 360 nm excitation signal that attained to a maximal value of 101 ± 1 arbitrary units at 250 s.
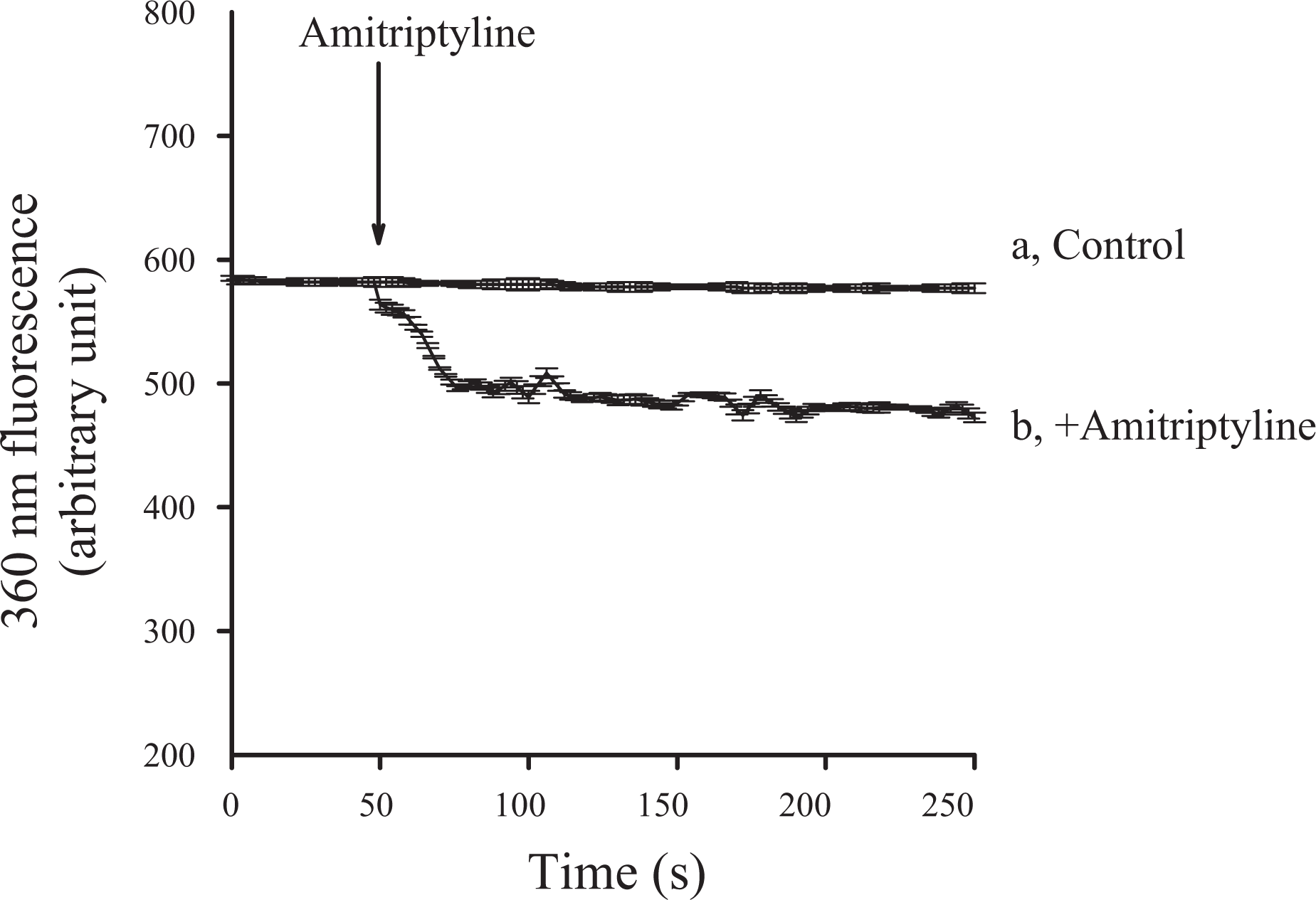
Effect of amitriptyline on Ca2+ influx by measuring Mn2+ quenching of fura-2 fluorescence. Experiments were performed in Ca2+-containing medium. MnCl2 (50 μM) was added to cells 1 min before fluorescence measurements. The y-axis is fluorescence intensity (in arbitrary units) measured at the Ca2+-insensitive excitation wavelength of 360 nm and the emission wavelength of 510 nm. Trace a: control, without amitriptyline. Trace b: amitriptyline (1000 μM) was added as indicated. Data are mean ± SEM of three separate experiments. SEM: standard error mean.
Regulation of amitriptyline-induced [Ca2+]i rises
Since amitriptyline concentration-dependently induced [Ca2+]i rises in the presence of Ca2+, the pathways of amitriptyline-induced Ca2+ entry were examined. Three Ca2+ entry inhibitors: nifedipine (1 μM), econazole (0.5 μM), and SKF96365 (5 μM); PMA (1 nM; a protein kinase C (PKC) activator); and GF109203X (2 μM; a PKC inhibitor) were added 1 min before amitriptyline (1000 μM) in Ca2+-containing medium. Figure 3 shows that except GF109203X, the other chemicals inhibited amitriptyline-induced [Ca2+]i rises by approximately 20%.

Effect of Ca2+ channel modulators on amitriptyline-induced [Ca2+]i rises. In blocker- or modulator-treated groups, the reagent was added 1 min before amitriptyline (1000 μM). The concentration was 1 μM for nifedipine, 0.5 μM for econazole, 5 μM for SKF96365, 10 nM for phorbol 12-myristate 13-acetate (PMA), and 2 μM for GF109203X. Data are expressed as the percentage of control (first column) that is the area under the curve (25–200 s) of 1000 μM amitriptyline-induced [Ca2+]i rises in Ca2+-containing medium and are mean ± SEM of three separate experiments. *p < 0.05 compared to the first column. Data are mean ± SEM of three separate experiments. Ca2+: calcium ion; PMA: phorbol 12-myristate 13-acetate; SEM: standard error mean.
Sources of amitriptyline-induced Ca2+ release
The endoplasmic reticulum has been established to be a dominant Ca2+ store in most cell types. 11,12 Thus, the involvement of the endoplasmic reticulum in amitriptyline-evoked Ca2+ release in MG63 cells was explored. To exclude the participation of Ca2+ entry, the experiments were performed in Ca2+-free medium. Figure 4(a) shows that addition of 1 μM TG, an endoplasmic reticulum Ca2+ pump inhibitor, 18 induced [Ca2+]i rises of 52 ± 2 nM. Addition of 1000 μM amitriptyline afterward induced tiny [Ca2+]i rises of 5 ± 2 nM. Figure 4(b) shows that after amitriptyline-induced [Ca2+]i rises, addition of thapsigargin failed to induce [Ca2+]i rises.
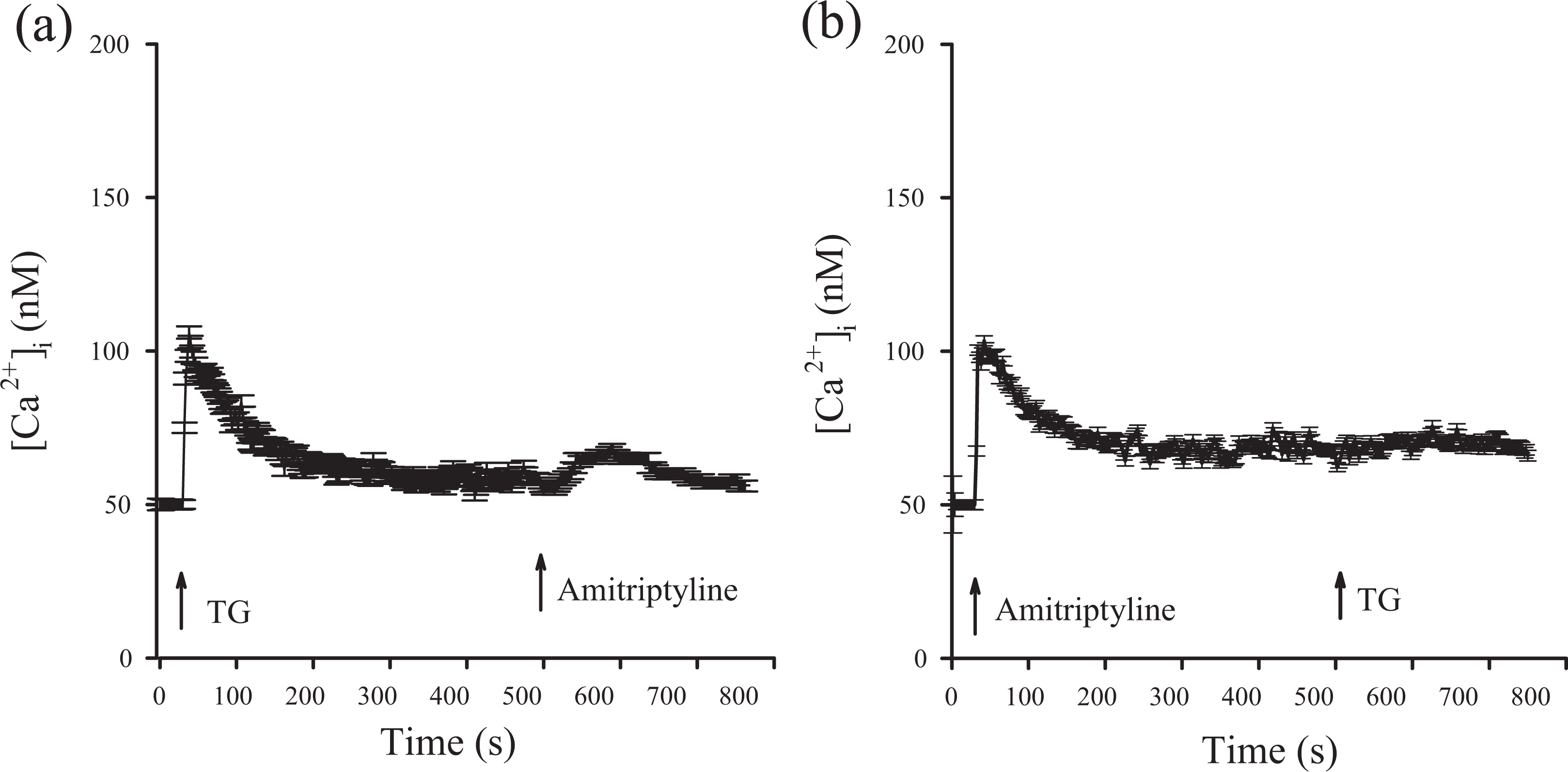
Effect of TG on amitriptyline-induced Ca2+ release. (a) and (b) TG (1 μM) and amitriptyline (1000 μM) were added at time points indicated. Experiments were performed in Ca2+-free medium. Data are mean ± SEM of three separate experiments. Ca2+: calcium ion; TG: thapsigargin; SEM: standard error mean.
Role of PLC in amitriptyline-induced [Ca2+]i rises
Among the numerous cellular regulatory enzymes, PLC is one of the pivotal proteins that modulate the release of Ca2+ from the endoplasmic reticulum. 11,12 Because amitriptyline released Ca2+ from the endoplasmic reticulum, the role of PLC in this response was examined. U73122, 19 a PLC inhibitor, was applied to see if the activation of this enzyme was needed for amitriptyline-induced Ca2+ release. First, Figure 5(a) shows that adenosine triphosphate (ATP) (10 μM) induced maximum [Ca2+]i rises of 48 ± 2 nM. ATP is a PLC-dependent agonist of [Ca2+]i rises in most cell types. 20 Second, Figure 5(b) shows that incubation with 2 μM U73122 did not alter basal [Ca2+]i but completely inhibited ATP-induced [Ca2+]i rises. The finding implicates that U73122 inhibited PLC activity. The results also suggest that incubation with 2 μM U73122 did not alter basal [Ca2+]i but inhibited 70% of 1000 μM amitriptyline-induced [Ca2+]i rises. U73343 (2 μM) is a U73122 analogue and was found to fail to inhibit ATP-induced effect (not shown).

Effect of U73122 on amitriptyline-induced Ca2+ release. Experiments were performed in Ca2+-free medium. (a) ATP (10 μM) was added as indicated. (b) From left to right (the time zone of the area under the curve), first column is 1000 μM amitriptyline-induced [Ca2+]i rises. Second column shows that 2 μM U73122 did not alter basal [Ca2+]i. Third column shows ATP-induced [Ca2+]i rises. Fourth column shows that U73122 pretreatment for 200 s completely abolished ATP-induced [Ca2+]i rises. Fifth column shows that U73122 (incubation for 200 s) and ATP (incubation for 50 s) pretreatment partially inhibited amitriptyline-induced [Ca2+]i rises. Data are mean ± SEM of three experiments. *p < 0.05 compared to first bar (control). Control is the area under the curve of 1000 μM amitriptyline-induced [Ca2+]i rises (25–190 s). Ca2+: calcium ion; ATP: adenosine triphosphate; SEM: standard error mean.
Cytotoxic effect of amitriptyline
Because unregulated [Ca2+]i rises may change cell viability, 11–12 experiments were performed to examine the cytotoxic effect of amitriptyline on MG63 cells. Cells were incubated with 0–500 μM amitriptyline for 24 h, and tetrazolium assay was conducted. Figure 6 shows that in the presence of amitriptyline, cell viability decreased in a concentration-dependent fashion between 200 μM and 500 μM. The question arose as to whether amitriptyline-induced cytotoxicity was caused by preceding [Ca2+]i rises. The intracellular Ca2+ chelator BAPTA/AM (5 μM) 21 was applied to prevent [Ca2+]i rises during amitriptyline incubation. Figure 6(a) shows that at a concentration of 1000 μM, amitriptyline did not evoke [Ca2+]i rises in BAPTA/AM-incubated cells. Figure 6(b) shows that 5 μM BAPTA/AM loading did not change control cell viability. In the presence of 200–500 μM amitriptyline, BAPTA/AM loading did not prevent amitriptyline-induced cytotoxicity.

Amitriptyline-induced Ca2+-independent cell death. (a) Following BAPTA/AM treatment, cells were incubated with fura-2/AM as described in section “Methods.” Then [Ca2+]i measurements were conducted in Ca2+-containing medium. Amitriptyline (1000 μM) was added as indicated. (b) Cells were treated with 0–500 μM amitriptyline for 24 h, and the cell viability assay was performed. Data are mean ± SEM of three separate experiments. Each treatment had six replicates (wells). Data are expressed as percentage of control that is the increase in cell numbers in amitriptyline-free groups. Control had 10,555 ± 714 cells per well before experiments and had 13,758 ± 715 cells per well after incubation for 24 h. *p < 0.05 compared to control. In each group, the Ca2+ chelator BAPTA/AM (5 μM) was added to cells followed by treatment with amitriptyline in Ca2+-containing medium. Cell viability assay was subsequently performed. Ca2+: calcium ion; BAPTA/AM: 1,2-bis(2-aminophenoxy)ethane-N, N, N′, N′-tetraacetic acid/AM; SEM: standard error mean.
Discussion
In terms of Ca2+ homeostasis research, amitriptyline was shown to increase [Ca2+]i in glioma cells 9 and CHO cells 10 but inhibit it in mouse trigeminal ganglion neurons. 6 The present study examined the effect of amitriptyline on Ca2+ homeostasis and the underlying mechanism in MG63 human osteosarcoma cells. The findings show that amitriptyline induced [Ca2+]i rises by depleting Ca2+ stores and causing Ca2+ influx. The data suggest that amitriptyline induced Ca2+ entry because the amitriptyline-induced Ca2+ response was suppressed by removal of extracellular Ca2+. Since removing extracellular Ca2+ decreased amitriptyline-induced response throughout the measurement period of 200 s, it implicates that Ca2+ entry occurred during the whole period. The ability of amitriptyline to induce Ca2+ influx was also independently demonstrated by amitriptyline-induced Mn2+ quench of fura-2 fluorescence.
The pathways underlying amitriptyline-induced Ca2+ entry was explored. Store-operated Ca2+ channels have been shown to play a key role in [Ca2+]i rises activated by several compounds in MG63 cells such as NPC-14686 13 and sertraline. 14 Nifedipine, econazole, and SKF96365 were applied to examine whether amitriptyline evoked Ca2+ entry through store-operated Ca2+ channels. 22 These three reagents have often been used to suppress store-operated Ca2+ entry in different cell types, even though there are no selective inhibitors so far. 23 –26 Our data show that all of these chemicals inhibited 20% of amitriptyline-evoked [Ca2+]i rises. Thus, it suggests that amitriptyline-induced Ca2+ entry might be via store-operated Ca2+ channels.
Ca2+ homeostasis has been shown to be affected by the activity of many protein kinases. 27 Our data show that amitriptyline-evoked [Ca2+]i rises were decreased by 20% when PKC activity was activated by PMA. In contrast, inhibition of PKC with GF109203X had no effect. This suggests that a normally regulated PKC level is necessary for amitriptyline to induce a full Ca2+ response. The relationship between store-operated Ca2+ entry and PKC has been well established in NPC-14686- or 3,3′-diindolylmethane-treated MG63 cells. 13,14 Thus, it appears that amitriptyline causes PKC-sensitive Ca2+ influx in MG63 cells.
The endoplasmic reticulum stores seemed to be the dominant one involved in amitriptyline-evoked Ca2+ release. The results show that incubation with the endoplasmic reticulum Ca2+ pump inhibitor TG inhibited 95% of amitriptyline-induced [Ca2+]i rises. In contrast, incubation with amitriptyline abolished TG-induced [Ca2+]i rises. The residual 5% might be contributed by other stores such as nucleus, cytosketolon, and so on. 11–12 The findings further show that the Ca2+ release was mediated by a PLC-dependent mechanism, because the release was inhibited by 70% when PLC activity was suppressed. Additional to PLC-dependent Ca2+ release, other Ca2+ release mechanisms may involve phospholipase A2 or NADPH oxidase. 28,29 Therefore, the mechanisms responsible for the residual 30% of amitriptyline-induced Ca2+ release in MG63 cells need further investigation.
Furthermore, our study shows that amitriptyline (200–500 μM) was cytotoxic to MG63 cells in a concentration-dependent manner. Ca2+ overloading is known to initiate processes resulting in changes in cell viability. 11,12 Because both [Ca2+]i rises and cell death were induced by amitriptyline in MG63 cells, it is crucial to understand whether the cytotoxicity occurred in a Ca2+-dependent fashion. Our findings show that 200–500 μM amitriptyline-induced cell death was not reversed under the condition that cytosolic Ca2+ was chelated by BAPTA/AM. This implies that amitriptyline-induced cell death was unrelated to [Ca2+]i rises. Although amitriptyline-induced Ca2+ signal did not cause cell death, it might interfere with numerous downstream Ca2+-sensitive processes such as malfunction of enzymes, genes, secretion, contraction, and so on 11,12 that integrate to alter physiology of MG63 cells. Therefore, it needs further assessment in the future research.
In our study, viability and [Ca2+]i measurements were performed under totally different conditions, thus the data cannot be compared. In cytotoxicity assays, cells were treated with amitriptyline overnight in order to obtain significant changes in viability. Although [Ca2+]i measurements were conducted online and terminated within 20 min, the cell viability was still >95% after 20 min incubation with amitriptyline. This explains why 200–500 μM amitriptyline decreased cell viability, whereas 1000 μM amitriptyline did not change viability in [Ca2+]i measurements.
In previous studies, amitriptyline at concentrations between 20 μM and 200 μM inhibited cell viability in myeloma and leukemia cells after treatment for 24, 48 or 72 h. 4 However, our study shows that 200–500 μM amitriptyline concentration-dependently inhibited cell viability in MG63 cells after treatment for 24 h. Therefore, the antitumor effect of amitriptyline on cytotoxicity may depend on cell types, treatment durations, and concentrations. Studies have been performed to explore the plasma level of amitriptyline in adults. The plasma level of amitriptyline may reach 20 μM. 30,31 This level may be expected to go much higher in patients with liver or kidney disorders or taking higher doses. In depression patients, the plasma concentration of amitriptyline after oral administration might be 10-fold higher than in healthy adults. 30,31 Our data show that amitriptyline at a concentration of 200 μM induced [Ca2+]i rises and cell death. Therefore, our results may be clinically relevant in some groups of patients. Previous studies suggested that because amitriptyline in the blood requires active transport into the liver, it is less metabolized by the cytochrome P450 family but exhibits more active renal excretion in adults. 30,31 Therefore, amitriptyline might have been excreted through kidney. 30,31 Furthermore, the antidepressant amitriptyline was shown to have potent therapeutic activity against multiple myeloma 32 and leiomyosarcoma. 33 Because this study shows that amitriptyline caused cytotoxicity in MG63 cells, the potential use of amitriptyline or its derivatives to cope with human osteosarcoma needs further exploration in the future.
Cortical spreading depression (CSD), reflected by alterations in Ca2+, Na+, and ATP-sensitive K+ channels, has been implicated in migraine and as a headache trigger. 34 A previous study has shown that amitriptyline may reduce CSD by affecting these ion channels. 34 Because amitriptyline affected Ca2+ homeostasis in rat brain cortex synaptosomes 5 and mouse trigeminal ganglion neurons, 6 this may contribute to strategies for CSD-associated migraine prophylaxis. Furthermore, psychiatric and neurological disorders are mostly associated with the changes in neural Ca2+ signaling pathways required for activity-triggered cellular events including neuron depolarization. 35 Among psychotropic drugs, amitriptyline seems to cause neuron depolarization and have protective effects on psychiatric disorders. 35 Therefore, investigation of the neuron depolarization in psychiatric disorders holds the promise of the development of amitriptyline.
Together, the results show that amitriptyline evoked [Ca2+]i rises by Ca2+ release from the endoplasmic reticulum in a PLC-dependent manner and Ca2+ entry via PKC-sensitive store-operated Ca2+ entry in MG63 human osteosarcoma cells. Furthermore, amitriptyline evoked cytotoxicity in a Ca2+-dissociated manner. Considering the [Ca2+]i-elevating and cytotoxic effects of amitriptyline found in MG63 osteosarcoma cells, caution should be exercised in using amitriptyline in other in vitro studies.
Footnotes
Authors’ Note
TL, CTC, and WZL contributed equally to this work.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by VGHKS104-097 to TL and VGHKS104-116 to C-RJ.