
Abstract
Airway epithelial damage and repair represents a novel therapeutic target in asthma and chronic obstructive pulmonary disease. An established mouse model of airway epithelial damage involves the Clara cell cytotoxicity of parenterally administered naphthalene, an important environmental toxicant with genotoxic and carcinogenic potential. The objective of the current study was to investigate naphthalene-induced toxicity and to identify and quantify DNA double-strand breaks in a murine naphthalene model of airway epithelial damage. Male C57/BL6 mice were injected with 200 mg/kg naphthalene and culled at 12-, 24-, 48- and 72-h time points. Lung function and bronchoalveolar lavage was performed and the lungs were dissected for histological analysis and for quantitation of DNA double-strand breaks using γH2AX as a molecular marker. Mice injected with naphthalene had increased epithelial denudation, bronchoalveolar lavage fluid cellularity and reactivity to nebulized methacholine chloride as compared to corn oil vehicle controls. Histological changes were most pronounced at the 12- and 24-h time points. DNA double-strand breaks, quantitated as the number of γH2AX foci per cell, were highest at the 24- and 48-h time points. All parameters had decreased at the 72-h time point, consistent with airway re-epithelization and cellular repair. Our findings indicate a time-dependent accumulation of γH2AX foci in mouse airway epithelial cells following administration of naphthalene. Naphthalene airway epithelial injury constitutes a model of DNA double-strand breaks in mice, which can be adapted as a suitable model for further investigation of genotoxic damage for evaluating the efficacy of potential therapeutics.
Introduction
Naphthalene (NA) is an aromatic hydrocarbon with the formula C10H8, consisting of two benzene rings. It is used as a household fumigant in the form of mothballs, is present in wood smoke, tar, asphalt and fossil fuels. It is also present in tobacco smoke. NA may cause haemolysis, blockage of renal tubules and hepatic necrosis in humans. 1 Individuals with hereditary glucose-6-phosphate dehydrogenase deficiency are particularly susceptible to NA-induced injury. There is some evidence from in vitro and animal models of genotoxicity and carcinogenicity.
Parenteral administration of NA to mice, hamsters and other animal species leads to dose-dependent airway epithelial cell necrosis.2,3 At 1–2 days after NA injection, Clara cells undergo cell death and exfoliation, leading to the appearance of debris in the airway lumen.4,5 This is of much importance, given that Clara cells are the progenitor cells of all of the cell types in the airway epithelium (ciliated, mucus-secreting goblet and surfactant secreting non-ciliated Clara cells). Overall, the NA administration protocol constitutes a model of airway epithelial damage with similarities to the pathology observed in asthma, chronic bronchitis and emphysema.
According to a previous liver study, many of the changes in cell organelles resemble those in Clara cells exposed to NA, that is, formation of large clear cytoplasmic vacuoles, dilation of smooth endoplasmic reticulum, loss of ribosomes and aggregation of microfilaments. It is proposed that intracellular glutathione (GSH) can protect cells from injury by scavenging reactive intermediates. 6 The epithelium of the distal conducting airways is one of the most susceptible sites for acute injury after toxicant exposure. The biochemical mechanisms of NA lung toxicity are well studied. Pulmonary toxicants (furans and hydrocarbons) are activated by the cytochrome P450 monooxygenase system in Clara cells. 7 Clara cells are located in large and small airways, containing electron-dense granules. They are thought to produce bronchiolar surfactant. In addition, they can metabolize xenobiotic compounds by the action of P450 enzymes.
The airway epithelium has attracted much interest recently, as it has a central role in asthma pathogenesis and damage to this structure in a chronic wound scenario contributes to both airway inflammation and remodelling. The bronchial epithelium acts as a physical and functional barrier, complemented by the mucociliary escalator, separating external insult and the internal milieu of the lung and therefore, provides the first line of airway defence.8,9 Physical damage of the epithelium allows tissue exposure to both exogenous and endogenous infectious and damaging agents which leads to airway wall structural changes (goblet cell hyperplasia, squamous metaplasia, disruption of tight junctions, loss of structural integrity, desquamation of epithelial cells and denudation of basement membrane [BM]) which will in turn lead to an inflammatory reaction and tissue damage. 10
In normal individuals, the self-repair process of the damaged epithelium is extremely fast and effective. 11 Injury usually causes the desquamation of cells from the epithelium (mainly columnar cells). Only minutes after the damage, neighbouring epithelial cells start to spread and migrate across BM and cover the denuded area—termed the restitution process. 12 In individuals with asthma, however, the airway epithelium is fundamentally abnormal in that it has an increased susceptibility to environmental infectious or toxic agents and an impaired self-repair process following damage. 13 Evidence suggests that this property of asthmatic epithelium is involved in the origin of asthma and is not just a consequence of excessive inflammation. The antioxidant pathways are defective and unable to generate antioxidant enzymes such as superoxide dismutase catalase and glutathione peroxidase, which are generally produced in non-asthmatic airways in order to protect airways from reactive oxygen species (ROS)-mediated tissue damage.13–15
The formation of clusters of ROS is known to induce DNA damage and amongst the different types of lesions DNA double-strand breaks (DSBs) are extremely lethal severely compromising genetic integrity.16,17 For example, it is known that ROS-mediated DSBs are formed in both cell culture systems (A549 cells) and in the epithelium following exposure to tobacco smoke exposure. 18 However, to our knowledge DSB induction has not been investigated in NA-induced lung injury models. Given the potent genotoxic effects of DNA DSBs, here we used γH2AX as a molecular marker, to investigate the formation of these lesions in a murine model of NA-induced airway epithelial damage.
Materials and methods
Reagents
Naphthalene and corn oil were purchased from Sigma Chemical Co. (St. Louis, MO, USA). NA was dissolved in corn oil as 20 mg/ml (100 mg NA in 5 ml oil), in a sterile environment (laminar flow hood) on the day of use.
Animals
Six- to eight-week-old male C57/BL6 mice (20–30 g) from the Walter and Eliza Hall Institute (Melbourne, Australia) were housed in the Murdoch Children’s Research Institute mouse facility under specific-pathogen free conditions and maintained on a 12-h light/dark cycle with food and water ad libitum for 4 days before use. Protocols used in this study were approved by the Murdoch Children’s Research Institute Animal Ethics Committee. Mice were injected with NA (200 mg/kg) intraperitoneally at 8 a.m. on day 1 or with corn oil (vehicle control, volumes were normalized for body weight).
Lung function assessment: Methacholine-induced airway hyperresponsiveness
Mice were randomly selected for the airway hyperresponsiveness (AHR) measurement at 12, 24, 48 or 72 h after exposure injection. AHR was induced by methacholine and measured by invasive plethysmography (Buxco Electronics, Troy, NY, USA). Tracheotomy was performed on mice anesthetised with ketamine (200 mg/g) and xylazil (10 mg/g) administered intraperitoneally. Animals were then placed in a plethysmograph chamber and ventilated with a small animal respirator (Harvard Apparatus, Holliston, MA, USA) at a rate of 120 strokes/min. A single dose of saline (baseline) and increasing concentrations of acetyl-β-methacholine ranging from 0.125 to 8 μg/kg (10 μl per delivery) were delivered by nebulizer administration. Maximal airway resistance and minimum dynamic compliance were measured (FinePointe; Buxco Electronics) for each dose. A dose-response curve of the difference between maximal resistance and baseline resistance against corresponding methacholine concentrations was obtained.
Bronchoalveolar lavage
Bronchoalveolar lavage (BAL) was collected after mice were culled by an overdose of ketamine (200 mg/g) and xylazil (10 mg/g) mix. A total of three, 0.5-ml lavages were pooled in ice-cold 20% fetal calf serum/phosphate buffered saline (FCS/PBS). BAL samples were then centrifuged (400 × g for 10 minutes) followed by removal of supernatant. Cell pellets were then resuspended in 5 ml of red blood cell lysis buffer for 5–7 minutes at room temperature (RT), followed by the addition of 2 ml 5% FCS/PBS. Samples were then centrifuged (500 × g for 5 minutes), washed twice with 5 ml 5% FCS/PBS, before resuspending in 0.5 ml 5% FCS/PBS. Total viable cell counts were determined using a haemocytometer and the trypan blue exclusion method (Sigma-Aldrich, St. Louis, MO, USA).19,20
Lung histopathology
The right lung lobe was dissected transversely into three sections, fixed in 10% neutral buffered formalin for 12–24 h and embedded in paraffin according to standard protocols. Tissue sections (3 μm) were randomly selected and stained. Histological changes were assessed in haematoxylin and eosin (H&E) stained sections. To differentiate between connective tissue and cytoplasm, lung sections were brought to water, immersed in Lilli Mayer haematoxylin and following washes with Scott’s tap water were incubated with Eosin Y (Grale Scientific, Melbourne, Australia). Excess stain was drained off and sections were dehydrated, cleared and mounted in DeePex mounting medium.
γH2AX immunofluorescence
Lung tissue, embedded in moulds in optimal cutting temperature (OCT) compound 4583, was sectioned (5 μm) and stained for γH2AX with primary rabbit polyclonal anti-γH2AX antibody (overnight incubation, diluted 1:500, in 1% BSA in PBS; Millipore) and a 1-h incubation with secondary antibody (Alexa Fluor 488 goat anti-rabbit IgG diluted 1:500, in 1% BSA; Invitrogen), as described in detail previously. 21 Nuclei were counterstained by mounting with Vectashield medium containing propidium iodide (PI).
The slides were sealed with nail varnish and stored overnight at 4°C in the dark before imaging. Images were acquired using a Z-series pattern (step size of 0.5 μm) with a Zeiss LSM510 meta confocal microscope (standard green fluorescent protein [GFP] for γH2AX—Alexa Fluor 488 goat anti-rabbit IgG and PI [543 nm] lasers were used).
Images were analysed using ImageJ (Fiji Version 1.44a) software. For quantitation of γH2AX foci, images were firstly split into their constituent colour channels (green—γH2AX and red—PI), a two-dimensional maximum intensity projection of both the γH2AX and PI channels was generated and the images were cleared of areas containing incomplete cells. A display showing γH2AX staining that occurs only within nuclei was generated by creating a mask from the PI projection, which was overlayed onto the γH2AX channel. To reduce background in the γH2AX channel, binary images were created and the PI maximum projection was multiplied by the γH2AX maximum projection. To count the foci, epithelial cells, identified with reference to H&E stained serial sections, were randomly selected and the noise tolerance value was adjusted so that the γH2AX foci were detected. Once identified using the vehicle control slide, the noise tolerance value was maintained constant for every image. Mean ± standard errors from six separate sections from two animals (>1000 cells counted per animal) were calculated.
Statistical analysis
Statistical significance was analyzed using the Student t test with Sigmaplot software (Version 10, Systat Software, Inc., Chicago, IL, USA).
Results
Naphthalene-induced airway epithelial damage
To investigate the characteristics of a mouse model of NA-induced acute epithelial injury, we compared epithelial denudation (epithelial injury), haematoxylin and eosin, BAL cell count and AHR (airway remodelling features) in the vehicle control group (CO) at NA-treated mice at four different time points (12-, 24-, 48- and 72-h post exposure). Total cell counts were performed on pooled BAL washes. NA damage resulted in increased numbers of lavage cells (p < 0.01) in the 24-h NA groups compared to the corn oil control group (Table 1 ).
Cell content of bronchoalveolar lavage of mouse lungs following NA administration

NA: naphthalene.
a p < 0.01 vs. untreated group.
H&E stained lung tissue sections from NA-treated and corn oil groups were examined to assess the impact of NA on airway epithelial denudation (Figure 1 ). Airway denudation was expressed as percentage of epithelial loss in each tissue section. While there was little or no denudation observed in the corn oil control group, the findings indicated that NA treatment led to significant epithelial denudation from as early as 12-h post injection (Figure 2 ; p < 0.05 versus corn oil control). The damage was most profound at 12 h (p < 0.001 versus corn oil control). The level of loss of integrity then gradually reduced over time to 24 h (p < 0.05 versus corn oil control), and 48 h (p < 0.01 versus corn oil control). At 72 h, damage persisted in the NA-treated group and the epithelium was not fully recovered (p < 0.05 versus corn oil control).

Haematoxylin and eosin stain of mouse airway following naphthalene (NA) administration. Following methacholine challenge and bronchoalveolar lavage, mouse lungs were dissected, fixed in formalin, processed and routine haematoxylin and eosin stain was performed. (A) The untreated vehicle control mouse airway epithelium is intact demonstrating no damage artefact from the tissue processing procedure. (B) Airway of mouse at 12 h post NA exposure shows significant detachment of airway epithelial cells. (C) Mouse airway at 24 h shows large area of denudation. (D) Mouse airway at 48 h showing area of localized epithelial damage. (E) At 72 h there is very little damage to the epithelium. Bar = 50 μm.

Quantification of epithelial cell damage in mouse airways following naphthalene (NA) administration. The percentage represents the proportion of damaged epithelium to the whole epithelium in the tissue section. Significant damage is found in all NA treatment groups compared to untreated vehicle only mice. *p < 0.05 and **p < 0.01. The mean percentage of damage is highest in the 12-h NA group and is reduced at subsequent time points.
Naphthalene administration causes a significant increase in airway hyperresponsiveness
Increasing concentrations of methacholine, a non-specific bronchodilator, was used to challenge the mice to test AHR and airway resistance as measured by invasive plethysmography. The mean maximal AHR is presented in Figure 3 . A marked increase in maximum resistance from baseline (saline) was observed for the NA-treated group at 12 h (p < 0.05) compared to the corn oil group. The significant difference between the two groups peaked at 24 h (p < 0.01). AHR for the NA-treated group returned back to baseline gradually over the subsequent 48- and 72-h time points.

Airway hyperresponsiveness of mice following naphthalene (NA) administration. Airway hyperresponsiveness was measured by invasive plethysmography. The maximal resistance following challenge with nebulized methacholine chloride was plotted. In the NA 12- and 24-h maximal resistance was significantly higher than in untreated vehicle only mice. *p < 0.05 and **p < 0.01.
Naphthalene induces DNA double-strand breaks in mouse lung epithelial cells
Our findings indicate a time-dependent accumulation of γH2AX foci in mouse epithelial cells following administration of NA (Figure 4 ). Quantitation of γH2AX foci indicate the number of foci increase from approximately 14 foci per cell in vehicle (untreated) mice to approximately 28 foci per cell 24 h following administration of NA (Figure 5 ). Maximal foci numbers (approximately 33 per cell) are observed 48 h following intraperitoneal (i.p.) injection of NA and this then decreases, but does not return to normal levels, at 72 h (Figure 5).
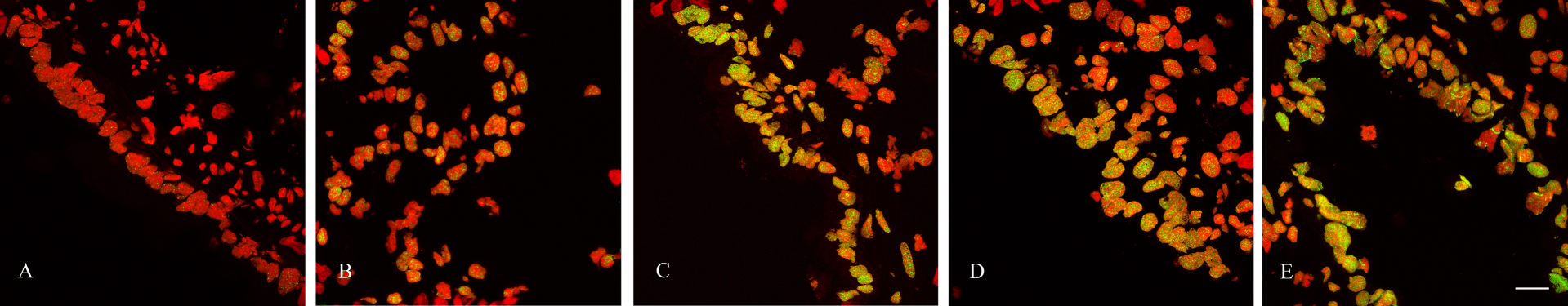
Accumulation of γH2AX foci in lung epithelial cells following naphthalene (NA) administration. Lungs were harvested from untreated vehicle only (A) and 12 (B), 24 (C), 48 (D) and 72 (E) hours following intraperitoneal (i.p.) administration of NA. Lung tissue, embedded in optimal cutting temperature (OCT) was sectioned (5 μm), fixed and stained for γH2AX (green). Nuclei were counterstained during mounting with Vectashield containing propidium iodide (PI; red). Images were acquired using a confocal microscope and the standard green fluorescent protein (GFP; for γH2AX—Alexa Fluor 488 goat anti-rabbit IgG) and PI (543 nm) lasers. Images were acquired in a Z-series pattern with a step size of 0.5 μm. Individual planes were deconvoluted and stacked to produce the merged maximum intensity images shown. Bar = 40 μm.

Quantitation of γH2AX foci in lung epithelial cells following naphthalene (NA) administration. Lungs harvested from untreated vehicle only and following intraperitoneal (i.p.) administration of 200 mg/ml NA for the indicated times were embedded in optimal cutting temperature compound 4583 and 5-μm sections were stained for γH2AX foci. Nuclei were counterstained during mounting with Vectashield containing propidium iodide (PI). Images were acquired with a confocal microscope using 0.5 μm Z-sectioning and foci were quantitated using ImageJ (Fiji Version 1.44a). Mean ± standard errors from six separate sections from two animals (>1000 cells counted per animal) are indicated (*p < 0.001).
Discussion
Acute naphthalene injury significantly damages the airway epithelium
Airway epithelial denudation is one of the most apparent markers of airway damage.22,23 NA was expected to cause this loss of epithelial integrity as exposure by i.p. injection route results in selective Clara cell damage due to cytochrome P450 produced reactive cytotoxic metabolites.8,24–26 Our findings indicated that NA treatment caused significant epithelial denudation with the difference between the NA and control groups being most profound at 12 h (Figure 2). The degree of loss of epithelial cells then gradually reduced over the time course. However, it still remained significant until 72 h, when compared to the corn oil control group (Figure 2). Previous studies have reported that damage of Clara cells are observed at 12 h where as complete exfoliation from the BM occurs 24 h after systemic NA exposure of the analogous dose used in this study. 5 It has also been reported that the airways are lined with a single layer of squamated cells with little to no associated inflammation at 24 h.5,27 Surviving epithelial cells then start to spread and migrate to cover the denuded area. This initial airway epithelial cell damage was followed by proliferation that resulted in a repaired airway epithelium by 10 days post exposure, with resolution and epithelial reconstitution completed by 20 days.1,5,28 Our findings are consistent with these previous studies.
Acute naphthalene injury has a significant impact on airway hyperresponsiveness
Our study provides evidence of an effect of NA on lung function via assessment of AHR by invasive plethysmography, which is the preferred technique of determining AHR in rodents. Based on previous observations, NA was expected to have an impact on AHR immediately after administration.27,28 We challenged with increasing doses of methacholine, and compared AHR between mice from NA-treated and corn oil control groups. The findings indicated that administration with NA does indeed cause a significant increase in AHR from as early as 12 h, peaking at 24 h. AHR dropped to baseline at the subsequent time points of 48 and 72 h. In a previous study, mid-expiratory airflow (EF50; measured in ml/s) was chosen as the parameter for testing lung function in the same strain of mice. In that study AHR was also induced with NA and non-invasive head-out plethysmography was used. In accordance with our findings, it was demonstrated that NA has a significant impact on AHR as early as 12 h post injection. 27
Genotoxicity of naphthalene
Polycyclic aromatic hydrocarbons, such as NA are well known to possess genotoxic and carcinogenic properties.29,30 For example, administration of inhaled NA in mice has been shown to result in increased nasal epithelial tumours in both sexes and in increase in alveolar adenomas and bronchial adenomas in females only. 31
Amongst the different types of DNA damage, DSBs are known to be one of the most critical lesions with respect to preservation of genomic integrity.16,17 Given the severity of DSBs mammalian cells have evolved elaborate signaling pathways to respond to these lesions.16,17,32 However, unrepaired or misrepaired DSBs (typically occurs when the error-prone non-homologous end-joining repair pathway is employed) are thought to be primary initiators of carcinogenesis. 17
It is well established that polycyclic aromatic hydrocarbons typically induce DSBs. 33 However, a previous study indicated that number of compounds except for NA resulted in the accumulation of γH2AX foci in a human keratinocyte cell. 34 Given this disparity, and to determine potential genotoxic effect, following histological examination of lung injury, we investigated whether NA administration induced DNA DSBs in mouse lung.
Phosphorylation of the histone variant, γH2AX on the Ser-139 residue forming γH2AX, is a well-established and highly sensitive marker of DNA DSBs.35–37 We used this phosphorylation event, as a molecular marker, to evaluate the effects of NA administration on DNA damage induction in mouse epithelial cells. The findings indicate accumulation of γH2AX foci in mouse epithelial cells following the administration of NA, which was time-dependent, with numbers observed at 24 and 48 h (Figures 4 and 5). Incidentally, frank DSBs were confirmed by co-localization studies with the DSB repair protein ataxia-telangiectasia mutated (ATM, data not shown). As indicated by the histological analysis, although the number of γH2AX foci per cell decreased significantly, epithelial damage and elevated DSBs persisted for up to 72 h.
Overall, our findings highlight the damaging properties and the ability of NA to induce AHR in mice. Further, our data highlights the genotoxic potential of NA. In this context, quantitation of DSBs using γH2AX as a molecular marker represents an extremely sensitive and specific tool that may be useful for further characterising NA-induced DNA damage and repair in mouse lung. Importantly, the method can be adapted for use in the evaluation of potential therapeutics that may circumvent the DNA damaging effects of NA.
Footnotes
Acknowledgements
The authors would like to acknowledge the technical and manuscript preparation assistance of Dr. Xizhe Fang and Miss Rosemary Gunawan.
The authors also acknowledge the grant and fellowship support from the Australian Institute of Nuclear Science and Engineering (AINSE), the National Health and Medical Research Council (NHMRC) and the CRC for Biomedical Imaging Development (CRC-BID).