
Abstract
Shikonin has been reported to regulate caudal-type homeobox 2 (CDX2)-mediated intestinal epithelial cell (IEC) differentiation, and ferroptosis has been identified a critical event during this process. However, the exact role of ferroptosis in shikonin-induced IEC differentiation remains unclear. Accordingly, the aim of this study was to elucidate the involvement of ferroptosis in CDX2-mediated IEC differentiation induced by shikonin. Real-time polymerase chain reaction, western blotting, luciferase assay, immunoprecipitation, and chromatin immunoprecipitation were used to reveal the mechanism underlying shikonin-modulated ferroptosis-dependent IEC differentiation in HT-29 and Caco-2 cells. Shikonin treatment reduced ferroptosis in IECs, as evidenced by the increased expression of glutathione peroxidase 4 (GPX4) and solute carrier family 7 (cationic amino acid transporter) member 11, which enhanced CDX2 expression and improved IEC barrier function. Mechanistically, shikonin activated the protein kinase A (PKA)/cAMP-responsive element-binding protein (CREB) signaling cascade, promoting CREB binding to the GPX4 promoter and initiating GPX4 transactivation. GPX4 inhibition reversed the effects of shikonin on CDX2 expression. Endogenous pyruvate kinase isozyme M2 interacted with phosphodiesterase 4; this interaction was disrupted by shikonin, leading to the activation of PKA/CREB signaling. The findings of this study indicate that a low dose of shikonin improves IEC barrier function through GPX4-mediated inhibition of ferroptosis, highlighting its potential as a therapeutic agent for intestinal mucosal injury.
Introduction
Impaired intestinal mucosa is a common pathological change in intestinal diseases such as Crohn’s disease, ulcerative colitis, and colitis-associated colorectal cancer.1,2 Studies have extensively elucidated the mechanisms underlying intestinal mucosal damage and potential therapeutic targets. Intestinal epithelial cell (IEC) differentiation is a critical process for recovering intestinal function, re-establishing intestinal mucosal integrity, and maintaining the balance between secretion and absorption. This differentiation is modulated by caudal-type homeobox 2 (CDX2), a key transcription factor that controls intestinal fate and maintains intestinal epithelial differentiation and barrier function by inducing the expression of mucin-2, lysozyme, sucrase-isomaltase, and villin.3–6 However, the mechanisms that regulate CDX2 expression have remained unelucidated.
The activation of ferroptosis, which plays a vital role in modulating gastrointestinal disease, has been recently observed in patients with inflammatory bowel disease (IBD).7,8 Ferroptosis, an iron-dependent form of regulated cell death driven by the lethal accumulation of membrane lipid peroxides, has been implicated in various diseases.9–11 For instance, kumatakenin ameliorates symptoms and suppresses intestinal inflammation in experimental colitis models by regulating the enolase-3/iron regulatory protein axis. 12 Deferasirox treatment reduces proinflammatory cytokine levels (interleukin (IL)-1β, IL-6, tumor necrosis factor (TNF)-α, and interferon (IFN)-γ) by inhibiting ferroptosis and reshaping the intestinal microbiota. 13 The expression of intestinal glutathione peroxidase 4 (GPX4), a primary antioxidant enzyme, is modulated by enterocytic differentiation, suggesting a protective role against oxidative damage in the gastrointestinal tract. 14 GPX4-mediated ferroptosis affects IEC differentiation,15,16 multidrug resistance, 17 and intestinal flora. 18 These findings suggest that ferroptosis inhibition may be critical for managing intestinal diseases. However, the regulatory mechanisms underlying ferroptosis remain poorly understood.
Targeting ferroptosis is a promising therapeutic strategy for improving various diseases. Recently, Shikonin (5,8-dihydroxy-2-[(1R)-1‑hydroxy-4-methylpent-3-enyl] naphthalene-1,4‑dione), a highly lipophilic naphthoquinone, isolated from the dried roots of Chinese herbs such as Lithospermum erythrorhizon, Onosma paniculata, and Arnebia euchroma,19,20 has demonstrated potential in treating various conditions, such as wound healing, inflammation, and cancer,21–23 particularly colitis. For instance, shikonin improves intestinal wound healing, 24 inhibits inflammatory cytokine release in dextran sulfate sodium-induced colitis models, 25 and modulates the T helper 17/regulatory T cell balance. 26 Although shikonin induces ferroptosis in various diseases at higher concentrations, low-dose shikonin triggers autophagy, characterized by increased LC3 expression, 27 and autophagy promotes ferroptosis. 28 The aim of this study was to elucidate the role of low-dose shikonin in IEC barrier function, providing comprehensive insights into its therapeutic potential.
Methodology
Reagents and antibodies
Roswell Park Memorial Institute (RPMI) 1640 medium (C11875500BT) and fetal bovine serum (FBS; 10099141) were purchased from Gibco (Waltham, MA, USA). RSL3 (HY-100218A), shikonin (HY-N0822), erastin (HY-15763), and ferrostatin-1 (HY-100579) were purchased from MedChemExpress (Monmouth Junction, NJ, USA) and dissolved in dimethyl sulfoxide (BL165B; Biosharp, Hefei, China). PrimeScript™ rt reagent kit (perfect real time; RR037A) and TB green® premix ExTaq™ II (Tli RNaseH Plus; RR820A) were obtained from Takara (Shiga, Japan). TRIzol reagent was purchased from Gibco (Waltham, MA, USA). Furthermore, the 4-Hydroxynonenal (4-HNE; SEKSM-0044) assay kit was purchased from Solarbio (Beijing, China). The protein A/G magnetic beads for immunoprecipitation (IP; B23202) were purchased from Selleck (Shanghai, China). The lipid peroxidation malondialdehyde (MDA) assay kit (S0131S), cell counting kit-8 (CCK-8; C0038), Lipo8000™ transfection reagents (C0533), and reactive oxygen species (ROS) assay kit (S0033S) were obtained from Beyotime (Shanghai, China). Duo-Lite luciferase assay system (DD1205-01) was purchased from Vazyme (Nanjing, China). A chromatin IP (ChIP) kit (magnetic, quantitative polymerase chain reaction (qPCR); ab270816) was purchased from Abcam (Cambridge, UK). Western Lightning® Plus-ECL (NEL105001EA) was purchased from Revvity (Shanghai, China). Nitrocellulose Blotting Membrane (10600001) was obtained from Cytiva (Shanghai, China). The shRNA plasmid targeting CREB (shCREB) was purchased from YouBio (Changsha, China). Pyruvate kinase isozyme M2 (PKM2)-specific polyclonal antibody (15822-1-AP), PKM2-specific monoclonal antibody (60268-1-Ig), GPX4 monoclonal antibody (67763-1-Ig), SLC7A11/xCT polyclonal antibody (26864-1-AP), CDX2 monoclonal antibody (26864-1-AP), Lamin A/C polyclonal antibody (10298-1-AP), beta actin monoclonal antibody (66009-1-Ig), cAMP responsive element binding protein (CREB) 1 polyclonal antibody (12208-1-AP), Phospho-CREB1 (Ser133) polyclonal antibody (28792-1-AP), beta actin monoclonal antibody (66009-1-Ig), phosphoglyceraldehyde dehydrogenase (GAPDH) monoclonal antibody (60004-1-Ig), alpha tubulin recombinant antibody (80762-1-RR), rabbit IgG control polyclonal antibody (30000-0-AP), mouse IgG (B900620), horseradish peroxidase (HRP)-conjugated AffiniPure goat anti-rabbit IgG (H + L; SA00001-2), and HRP-conjugated AffiniPure goat anti-mouse IgG (H+L; SA00001-1) were purchased from Proteintech (Wuhan, China). Anti-phosphodiesterase 4 (PDE4; ab14628) and anti-PDE4D (ab99409) antibodies were sourced from Abcam (Cambridge, UK). Phospho-protein kinase A (PKA) C-alpha (PRKACA)-T197 rabbit pAb (60243-1-Ig) and PKA C-alpha (PRKACA) mouse mAb (A21869) were obtained from Abclonal (Wuhan, China). Ultrapure reagents were purchased from Biosharp (Hefei, China).
Cell culture, treatment, and transfection
HT29 and Caco-2 cells were purchased from ATCC (Manassas, VA, USA) and cultured in RPMI-1640 medium supplemented with 10% FBS. Cells were treated with 0.05 μM shikonin, and untreated cells served as the control group. Additionally, cells were treated with RSL3 (2 μM), erastin (5 μM), and ferrostatin-1 (Fer-1, 10 μM). shCREB and reporter plasmids were constructed by Youbio (Changsha, China) and delivered into cells using lipo8000™ transfection reagents following the manufacturer’s instructions.
qPCR analysis
The total RNA was extracted using TRIzol reagent following the manufacturer’s instructions. Reverse transcription and qPCR were performed to assess the mRNA expression of the indicated genes using Primescript™ RT reagent kit (RR037A) and TB Green® premix ExTaq™ II (RR820A). The following RT-qPCR primers were used: β-actin forward 5′-CACCATTGGCAATGAGCGGTTC-3′ and reverse 5′-AGGTCTTTGCGGATGTCCACGT-3′; CDX2 forward 5′-CTCGGCAGCCAAGTGAAAACCA-3′ and reverse 5′-GCTTTCCTCCGGATGGTGATGTA-3′; GPX4 forward 5′-GAGGCAAGACCGAAGTAAACTAC-3′ and reverse 5′-CCGAACTGGTTACACGGGAA-3′; SLC7A11 forward 5′-ACGGTGGTGTGTTTGCTGTCTC-3′ and reverse 5′-GCTGGTAGAGGAGTGTGCTTGC-3′.
Subcellular fractionation isolation and immunoblotting analysis
Subcellular fractionation was performed using a nuclear and cytoplasmic protein extraction kit (P0027; Beyotime, Shanghai, China). Total protein was harvested using a 2× loading buffer and separated after treatment. Briefly, proteins were transferred to a nitrocellulose membrane and blocked with 5% milk for 1 h. The membrane was then incubated with the primary antibodies overnight at 4 °C to detect the indicated protein bands. After washing with phosphate‑buffered saline with Tween 20 for 15 min, secondary antibodies were added, and the membrane was incubated for 1 h. Protein bands were detected and imaged using Western Lightning® Plus-ECL.
Immunofluorescence
Briefly, after treatment, the cells were fixed, permeabilized, and incubated with CDX2 antibody overnight at 4 °C. Subsequently, samples were incubated with an Alexa Fluor 488-conjugated secondary antibody for 1 h. Coverslips were mounted on glass slides using ProLong™ gold antifade reagent containing 4′,6‑diamidino‑2‑phenylindole, and the stained cells were imaged under a laser-scanning confocal fluorescence microscope.
ROS detection
ROS levels were measured using 2′,7′‑dichlorodihydrofluorescein diacetate (DCFH-DA) staining following the manufacturer’s instructions. Briefly, the cells were seeded into a 12-well plate and treated with or without shikonin for the indicated time. After washing with phosphate-buffered saline (PBS), DCFH-DA solution was added, and the cells were incubated for 20 min at 37 °C in 5% CO2. ROS levels were visualized and captured.
CCK-8 analysis
Cell viability was assessed using the CCK-8 assay following the manufacturer’s instructions. Briefly, the cells were digested, reseeded into a 96-well plate, and allowed to adhere overnight. After treatment with various concentrations of shikonin for 48 h, the medium was replaced with fresh medium containing 10% CCK-8 reagent and incubated for 30 min. The absorbance was measured at 450 nm to determine cell viability. Each concentration was tested with six biological replicates.
MDA assay
MDA levels were measured following the manufacturer’s instructions. Briefly, the cells were incubated with cell lysis buffer for western blotting and IP (P0013) for 10 min to obtain the supernatant. Subsequently, 100 μL of the sample was mixed with 0.2 mL of the working solution, and the mixture was incubated in a water bath at ⩾95 °C for 20 min. After cooling, the samples were transferred to a 96-well plate, and the absorbance was measured at 530 nm using a microplate reader.
4-HNE assay
The 4-HNE levels were measured following the manufacturer’s instructions. After preparing the cell lysates, 50 μL of the sample was added to each well in a 96-well plate, followed immediately by 50 μL of the biotin-antibody working solution. The plate was incubated at 37 °C for 45 min and washed thrice. Subsequently, 100 µL of the enzyme conjugate working solution was added to each well. Next, the plate was covered with a sealing film and incubated at 37 °C for 30 min. After washing the plate five times, 90 µL of the substrate was added to each well. The wells were covered with a sealing film and incubated at 37 °C in the dark for 15 min. Furthermore, 50 µL of the stop solution was added, and the optical density of each well was measured at 450 nm.
Proximity ligation assay (PLA)
PLA was performed as previously described. 29 Briefly, the molecular interaction between PKM2 and PDE4 was evaluated using a Duolink® In Situ Red Starter Kit Mouse/Rabbit (Sigma–Aldrich) according to the manufacturer’s instructions. Cells were treated with or without shikonin for 1 h in an eight-well cell culture chamber slide (07-2108; Biologix). Subsequently, the cells were fixed in 4% paraformaldehyde at room temperature for 30 min and stained with anti-PKM2 and anti-PDE4 antibodies at 4 °C overnight. After washing, the cells were incubated with the PLA probe mixture and the ligation solution at 37 °C. For amplification, a polymerase solution containing fluorescence-labeled oligonucleotides was applied for 90 min at 37 °C. Fluorescent spots and images were captured using a Zeiss LSM 700 confocal microscope.
IP
After treatment with or without shikonin for 1 h, the total protein was extracted using IP lysis buffer. The lysates were incubated with anti-PKM2 antibody overnight at 4 °C. Protein A/G beads were added, and the sample was incubated for 1 h at 4 °C to pull down immune complexes. After washing, the immunoprecipitated complexes were eluted and examined using immunoblotting.
Luciferase analysis
The reporter plasmid containing the GPX4 promoter, combined with the Renilla plasmid (internal control), was transfected into cells using Lipo8000™ transfection reagent for 24 h. Cells were treated with or without shikonin for 24 h. The Renilla value served as the internal control. The relative luciferase unit, calculated as firefly/Renilla, was measured using the Duo-Lite luciferase assay system.
ChIP
Cells were grown to 80% confluence in a serum-free medium overnight and treated with or without shikonin for 1 h. ChIP experiments were performed following the manufacturer’s instructions. Cross-linking was performed with an incubation solution containing fixation buffer and formaldehyde for 10 min with gentle shaking, and stopped by the addition of glycine. Subsequently, the cells were washed with PBS and harvested by scraping. Centrifugation was performed to remove the supernatant, and the lysis buffer was added and incubated for 10 min at 4 °C with gentle mixing to isolate cell nuclei. After further incubation with the complete shearing buffer, IP with the indicated antibodies was performed using sheared chromatin. Real-time PCR was used to amplify the GPX4 promoter sequence with specific primers, as previously described by Pan et al. 15
Statistical analysis
Statistical analyses were performed using GraphPad Prism V software. Each experiment was performed with at least three biological replicates. Results with p < 0.05 were considered statistically significant.
Results
Shikonin induced CDX2 expression
To explore the effect of shikonin on IEC differentiation, we used Caco-2 cells as a model system. CCK-8 analysis was performed to determine the optimal shikonin concentration for the experiments. As shown in Figure 1a, low shikonin doses exhibited no significant cytotoxicity, whereas high doses (>2.5 μM) inhibited cell viability. Moreover, shikonin protected the barrier function of IECs by enhancing transepithelial electrical resistance in a dose-dependent manner (Figure 1b). Therefore, we selected 0.05 μM shikonin for further experiments. Further analysis revealed that shikonin stimulation increased CDX2 mRNA and protein levels in Caco-2 cells (Figure 1c and d). In addition, immunofluorescence analysis showed enhanced CDX2 expression in HT29 cells after shikonin treatment (Figure 1e).
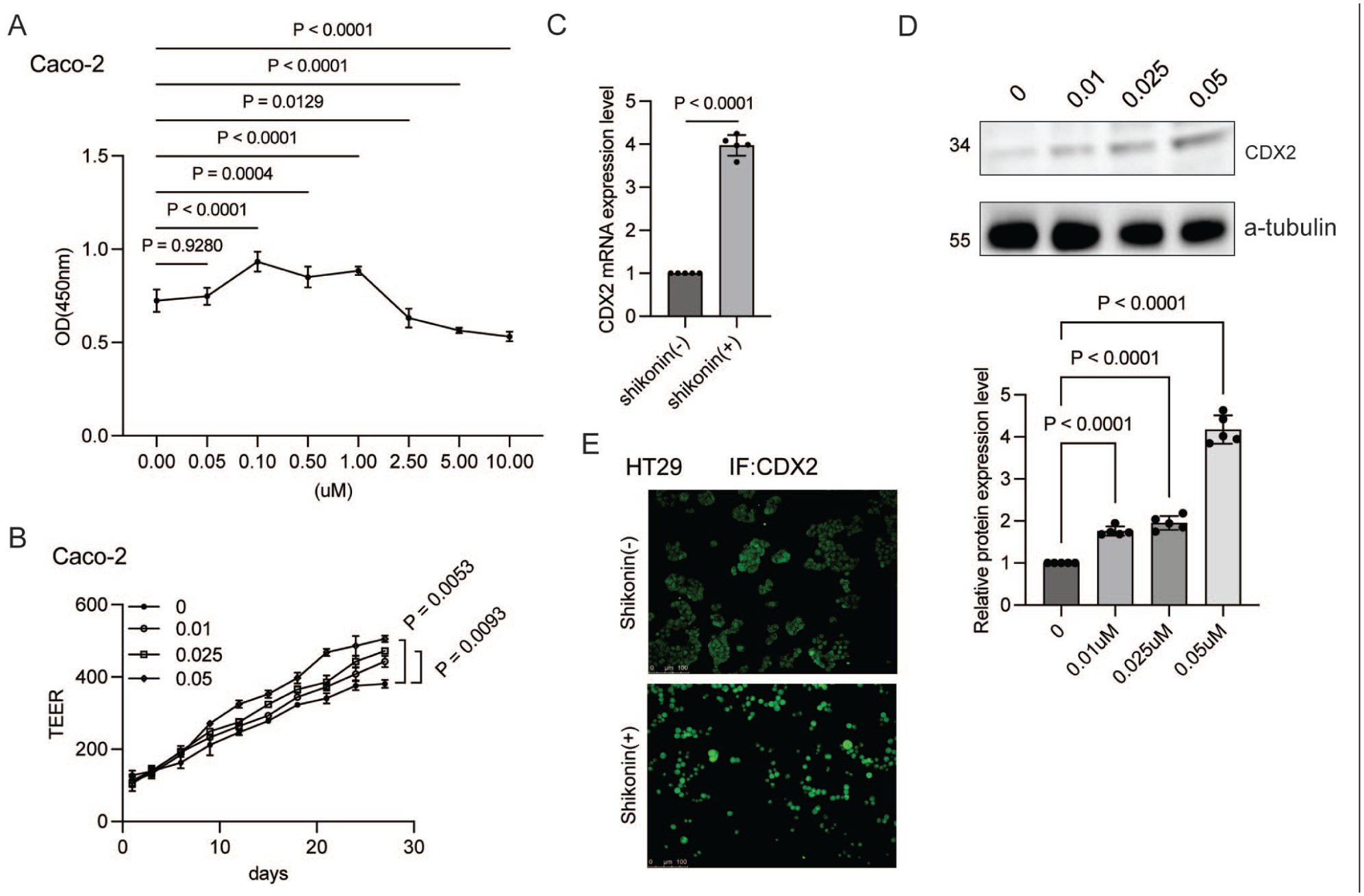
Shikonin induces CDX2 expression and enhances barrier function. (a) CCK-8 analysis of shikonin cytotoxicity at various concentrations in Caco-2 cells after treatment for 48 h (n = 5). Data are presented as mean ± SD and determined using a one-way ANOVA. (b) The effect of shikonin on TEER over time (n = 3). A one-way ANOVA was used to evaluate the difference. Data are displayed as mean ± SD. The control group represents cells that were not treated with shikonin. (c) qPCR analysis of CDX2 expression in Caco-2 cells treated with or without shikonin for 48 h. The difference was determined using a one-sample T-test (n = 5). (d) Immunoblotting reveals CDX2 expression in Caco-2 cells treated for 48 h. The band intensity was quantified and analyzed using a one-way ANOVA (n = 6). Data are presented as mean ± SD. (e) IF analysis of CDX2 expression in HT29 cells treated with or without shikonin for 48 h. Scale bar: 100 μm.
Shikonin regulated CDX2 expression through ferroptosis
Studies have shown that ferroptosis inhibition is closely associated with cell differentiation14,15; however, whether ferroptosis is involved in shikonin-mediated regulation of CDX2 in IECs remains unknown. Real-time PCR and western blotting showed that shikonin treatment substantially upregulated GPX4 expression at the mRNA and protein levels (Figure 2a and b). Moreover, MDA, ROS, and 4-HNE levels were significantly reduced by shikonin treatment (Figure 2c–e), suggesting that shikonin suppressed ferroptosis. Subsequently, we determined whether ferroptosis activation could reverse the effect of shikonin on CDX2 expression. Treatment with RSL3 or erastin, ferroptosis inducers, reversed the increase in CDX2 expression after shikonin stimulation (Figure 2f), whereas the ferroptosis inhibition by ferrostatin-1 aggravated shikonin-induced CDX2 expression (Figure 2g). These data suggest that shikonin suppresses GPX4-mediated ferroptosis and induces CDX2 expression.

Shikonin regulates ferroptosis. (a) After stimulating Caco-2 cells with or without shikonin for 48 h, the total RNA was extracted to assess GPX4 and SLC7A11 mRNA expression levels. Data are displayed as mean ± SD and determined using a one-sample T-test (n = 5). (b) The cells were treated as described in (a), and western blotting was performed to assess GPX4 and SLC7A11 protein levels in Caco-2 cells. Band intensities were quantified and analyzed using a one-sample T-test. Data are presented as mean ± SD (n = 5). After treatment with or without shikonin for 48 h, MDA (c), ROS (d), and 4-HNE (e) levels were determined according to the manufacturer’s instructions. Data are presented as mean ± SD and analyzed using one-sample T-tests (n = 4). (f, g) After treatment as indicated for 48 h, CDX2 and GPX4 levels were determined using immunoblotting.
Shikonin regulated ferroptosis through CREB
Recent studies demonstrated that GPX4 is a direct target of CREB.14,30,31 Thus, the shikonin-induced inhibition of GPX4-mediated ferroptosis was attributed to CREB. A reporter gene containing the GPX4 promoter was constructed and transfected into HEK293 cells along with an internal Renilla plasmid (pGL4.74), shCREB, or empty vector for 12 h, followed by shikonin stimulation for 36 h. Shikonin treatment significantly upregulated GPX4 luminescence, whereas CREB depletion by shCREB reversed the shikonin-induced increase in GPX4 promoter activity; CREB expression levels confirmed the knockdown efficiency (Figure 3a). In addition, ChIP analysis revealed that stimulation with shikonin notably enhanced the ability of CREB to bind to the GPX4 promoter (Figure 3b). Notably, CREB knockdown in HT29 cells abolished the shikonin-mediated upregulation of GPX4 and CDX2 (Figure 3c). These results suggest that shikonin suppresses GPX4-mediated ferroptosis in a CREB-dependent manner.

CREB regulates shikonin-modulated GPX4 expression. (a) Luciferase reporter assay of GPX4 promoter activity in Caco-2 cells co-transfected with pGL4.74-GPX4 reporter gene and empty vector or shCREB for 24 h, followed by treatment with or without shikonin (0.05 μM) for 24 h. Data are presented as mean ± SD and analyzed using a two-way ANOVA (n = 5). Western blotting confirmed the knockdown efficiency. (b) After stimulation with or without shikonin for 1 h, Caco-2 cells were fixed and subjected to nuclear fractionation. Chromatin immunoprecipitation was performed using anti-CREB or IgG. qPCR was used to examine the effect of shikonin on CREB binding to the GPX4 promoter. Data are shown as mean ± SD and analyzed using a two-way ANOVA (n = 5). (c) Caco-2 cells were transfected with or without shCREB for 12 h, followed by treatment with or without shikonin for 48 h. The total protein was collected to assess the expression of the indicated proteins. The band density was analyzed using a one-way ANOVA. Data are presented as mean ± SD (n = 4).
Shikonin modulated CREB activation and nuclear translocation
The above findings suggest that CREB is crucial for shikonin-mediated GPX4 expression. Because CREB phosphorylation is a critical event for its nuclear translocation, which initiates target gene transactivation,15,32 we explored the influence of shikonin on CREB phosphorylation and nuclear translocation. Shikonin treatment increased nuclear CREB levels. Histone 3 and tubulin served as internal controls for the nuclear and cytosolic fractions, respectively (Figure 4a). Moreover, further results showed that shikonin regulated the phosphorylation of PKA and CREB within the PDE4/PKA/CREB signaling pathway, causing CREB nuclear translocation, whereas no significant changes in baseline PKA/CREB expression were observed (Figure 4b). Notably, Co-IP analysis revealed that endogenous PKM2 interacted with PDE4; this interaction was inhibited by shikonin treatment (Figure 4c). Consistent with this, PLA showed that the interaction between PKM2 and PDE4 was reduced after shikonin treatment (Figure 4d). These findings suggest that shikonin triggers CREB activation and nuclear translocation via PDE4/PKA signaling, inducing GPX4 transactivation and increasing CDX2 expression.

Shikonin modulates PDE4/PKA/CREB activation and induces CREB nuclear translocation. (a) The cell fraction of the indicated group was isolated to detect CREB levels in Caco-2 cells treated with or without shikonin for 1 h; histone 3 and alpha-tubulin served as the internal control for nuclear and cytosolic protein, respectively. (b) The total protein was harvested to detect the indicated proteins in Caco-2 cells treated with or without shikonin for 1 h. (c) Co-IP analysis of PKM2 and PDE4 complex formation after shikonin treatment for 1 h. (d) PLA was performed to detect the relationship between PKM2 and PDE4.
Discussion
To the best of our knowledge, this study is the first to demonstrate that a low dose of shikonin can trigger PKM2/PDE4/PKA/CREB-mediated GPX4 expression, suppressing ferroptosis and improving intestinal barrier function in IECs. The activation of ferroptosis by the GPX4 inhibitor RSL3 reversed the effect of shikonin on CDX2 expression. Notably, shikonin treatment disrupted the interaction between PKM2 and PDE4, inhibiting PDE4 phosphorylation, activating PKA/CREB signaling, and enhancing CREB nuclear translocation. This study reveals a novel mechanism by which shikonin promotes IEC differentiation in a ferroptosis-dependent manner, providing more comprehensive insights into the effects of shikonin at low doses.
Huang et al. showed that a high dose of shikonin (>2 μM), but not a low (<1 μM) dose, could reduce IL-1β, IL-6, and TNF-α expression in macrophages stimulated by lipopolysaccharides and IFN-γ, inhibiting M1 macrophage polarization in the colonic lamina of ulcerative colitis mice.33,34 Moreover, shikonin at concentrations <1 μM does not significantly inhibit the activation of NLRP3 inflammasomes, 35 whereas at 1 μM, it significantly enhances IEC restitution in vitro without affecting cell proliferation. 24 In addition, different doses of shikonin have opposing effects on NRF2 expression.36,37 In the present study, we found that low doses of shikonin improved intestinal epithelial barrier function by inhibiting ferroptosis, whereas high doses reportedly promote ferroptosis. We will explore the mechanism underlying these dose-dependent contrasting effects in future studies.
cAMP is synthesized by adenylyl cyclase (AC), which converts ATP to cAMP upon activation of Gαs-protein coupled receptors. cAMP is further converted to 5′-adenosine monophosphate by PDE. Classical cAMP signaling, regulated by AC-induced cAMP synthesis and PDE4-mediated cAMP hydrolysis, activates PKA phosphorylation, triggering the phosphorylation of transcription factors and initiating the expression of target genes, including CREB, ATF3, KLF11, NRF2, and Smad3.15,38–41 Targeting PDE4 can markedly alleviate various diseases, including IBD and lung fibrosis.42,43 In the present study, PKM2 inhibition by shikonin disrupted the PKM2/PDE4 complex, decreasing PDE4 phosphorylation and triggering PKA/CREB signaling to initiate GPX4 transactivation. However, further studies are required to identify the critical PKM2 domain that interacts with PDE4 to regulate PDE4 phosphorylation. In addition to CREB, our present findings cannot completely rule out the possibility that shikonin mediates GPX4 expression through other transcription factors. The potential role of shikonin in AC phosphorylation would be explored in our future studies. Moreover, there are several limitations in this study, including lacking in vivo experiments to confirm in vitro results and the changes of dimers/tetramers formation PKM2 status under low doses of shikonin, which would be addressed in our next work. taken together, current results provide novel insights into shikonin-regulated IEC barrier function.
Conclusion
Our study demonstrates that a low dose of shikonin triggers GPX4 expression and inhibits ferroptosis, enhancing intestinal barrier function. These findings provide insights into optimizing shikonin-based interventions and improving therapeutic strategies for restoring intestinal mucosal integrity in intestinal disorders.
Footnotes
Author contributions
FHY and ZL conceptualized the study; FHY, PHL, HLG, WZY, and GW curated the data and performed the investigation; FHY, PHL, GW, and XFT formally analyzed the data; PHL, HLG, WZY, and XFT determined the methodology; ZL supervised the study and administered the project; FHY and PHL wrote, edited, and reviewed the manuscript. All the authors have read and approved the final version of the manuscript.
Declaration of conflicting interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This study was supported by a Traditional Chinese Medicine grant from Guangdong (20232158).
Ethics approval
Not applicable.
Informed consent
Not applicable.
Data availability statement
The datasets generated and/or analyzed in the current study will be made available by the corresponding author upon reasonable request.