
Abstract
Mice homozygous for a dominant-negative allele of the Clock gene (ClockΔ19/Δ19) have slightly but significantly decreased male fertility. The molecular mechanism for this reduction in fertility is unknown. In the present study, we used a small hairpin RNA (shRNA) strategy to specifically knock down the Clock gene expression in the testes of male mice and determined its effect on male fertility. Clock knockdown led to smaller litter size, a lower in vitro fertility rate, lower blastula formation rate, and lower acrosin activity of the knockdown sperm. Locomotor activity analysis of the Clock knockdown mice revealed that Clock knockdown in testes did not alter their circadian rhythm. Taken together, these results provide the first evidence that Clock gene expression in round spermatids is essential for maintaining male reproductivity and suggest that acrosin may be a novel regulatory target of the Clock gene that would regulate the fertilization and early embryonic development to blastula. These findings may provide new clues for development of novel male contraceptive strategies.
Virtually all living organisms have circadian rhythms that regulate their metabolic and behavioral rhythms and are synchronized to environmental conditions such as light-dark cycles and temperature variations (Lim et al., 2006). Clock, a core component of the circadian system, was discovered by forward mutagenesis using N-ethyl-N-nitrosourea and behavioral screening for circadian rhythm abnormalities (Vitaterna et al., 1994).
Reproduction is an activity that is strongly influenced by a circadian system in most organisms (Goldman, 1999; Turek, 1992; Turek et al., 1984); it has been widely investigated in insects (Beaver and Giebultowicz, 2004; Beaver et al., 2002, 2003; Giebultowicz and Riemann, 1990; Johnson and Day, 2000). In mammals, circadian clock genes are expressed in many peripheral organs (Tei et al., 1997). Oscillations in peripheral tissues are synchronized to environmental cycles by the master regulator of the circadian system, the suprachiasmatic nucleus (SCN) of the hypothalamus (Yoo et al., 2004). An earlier study showed that the litter size generated by homozygous ClockΔ19/Δ 19 mutant males is about 21% lower than that for wild-type male mice (Dolatshad et al., 2006). This reduction in fertility was attributed to the altered circadian rhythms in ClockΔ19/Δ 19 mutant mice (Dolatshad et al., 2006). However, expression of the circadian genes in mammalian testes, including Per1, Per2, Bmal1, Clock, and Cry1, is constant, suggesting that these clock proteins have noncircadian functions in spermatogenesis (Alvarez et al., 2003). By studying circadian mutant or knockout mice, it is difficult to determine whether phenotypic alterations are due to changes in circadian rhythms or to alterations of gene expression level that may not be related to circadian rhythms (e.g., transcription factor role). Circadian clock genes are expressed during spermatogenesis in a stage-specific and circadian-independent manner (Alvarez et al., 2003; Bittman et al., 2003; Morse et al., 2003). Clock gene expression has been detected exclusively in round spermatids (Alvarez et al., 2003). It takes about 14 days for round spermatids to differentiate into the species-specific shaped spermatozoon (Clermont and Trott, 1969; Fujii et al., 2002; Hecht, 1998).
In this study, we chose the small hairpin RNA strategy to specifically knock down Clock gene expression in the testes of male mice; we determined its effect on male reproduction and investigated the underlying mechanisms.
Materials and Methods
Plasmids
Clock small hairpin RNA expression plasmid (pClock.shRNA) and its control (pCtrl.shRNA) were purchased (Genesil Biotechnology Co., Ltd., Wuhan, China). The entire DNA sequence inserted in pGensil-1 plasmid to construct pClock.shRNA was 5′-GGATCCGCTTCAGACTCATTATTATTTCAAGACGATAATAATGAGTCTGAAGCTTTTTTGTCGACAAGCTT-3′ including the restriction enzyme cutting sites of BamHI and HindIII, and its expression was driven by hU6 promoter. The inserted sequence was verified by sequencing.
Animal Husbandry and Locomotor Activity Monitoring
All studies were performed in sexually mature mice (8-10 weeks in male mice, 6-8 weeks in females) of the ICR strain (Mus musculus). All animal procedures were approved by the Ethical Committee of Investigation of Laboratory Animals of Sichuan University. Male mice for locomotor activity monitoring were kept under a constant dark condition. These mice were kept individually in plastic cages (45 × 35 × 30 cm) that were equipped with a running wheel (15-cm diameter). Infrared switches attached to the running wheels were connected to a data acquisition system that recorded the number of wheel revolutions in 3-min bins. Male mice for other tests and female mice were kept in cages on a 12-h light/12-h dark lighting schedule. All mice had food and water available ad libitum.
Plasmid Transfection
The shRNA expression plasmid was injected into the testes of the sexually mature male mice using an in vivo jetPEI (Polyplus, Illkirch Cedex, France) according to the manufacturer’s instructions. Briefly, mice were anesthetized with diethyl ether and both testes were injected with 20 µL of the plasmid-liposome complex using a microinjector (32 Ga).
Immunohistochemistry and Western Blot
Male mice were anesthetized with an intraperitoneal injection of 40 mg/kg pentobarbital and their testes were surgically removed on the 3rd, 6th, and 18th day of the siRNA plasmids injection, respectively, 3 mice on each day. One testis of each mice was fixed in 4% paraformaldehyde, paraffin embedded, and sectioned (thickness of 5 µm). Slide-mounted sections were then placed in xylene to remove the paraffin wax and incubated in decreasing concentrations of ethanol and then tap water to rehydrate the tissue, followed by incubation in 2% hydrogen peroxide to quench the endogenous peroxidase activity and washing in PBS. Then the sections were heated in 10 mM sodium citrate buffer (pH 6.0) for antigen retrieval. Subsequently, samples were blocked with normal goat serum and then incubated with primary antibody (1:100 for anti-CLOCK; Santa Cruz Biotechnology Inc., Santa Cruz, CA, USA) overnight at 4 °C. Following 3 washes in PBS, sections were incubated with horseradish peroxidase (HRP)–conjugated goat anti-mouse secondary antibody for 1 h at room temperature. Immunoreactivity was visualized with diaminobenzidine (DAB) and counterstained with Mayer’s hematoxylin. Coverslips were applied, and the slides were viewed using a light microscope.
The other testis of each treated mice was harvested and homogenized. Sperms were collected from the cauda epididymis of mice on 18th day of the siRNA plasmids injection. Proteins were extracted with RIPA Lysis Buffer (Beyotime Institute of Biotechnology, Shanghai, China) according to the manufacturer’s instructions. Protein concentration was determined by Enhanced BCA Protein Assay Kit (Beyotime Institute of Biotechnology) according to the manufacturer’s instruction. Proteins were separated by 10% SDS-polyacrylamide gel electrophoresis, followed by blotting onto membranes. Blots were probed with an anti-CLOCK monoclonal antibody (Santa Cruz Biotechnology) and an HRP-labeled goat anti-mouse IgG antibody (ZSGB-BIO, Beijing, China) for detecting CLOCK and with an anti-ACROSIN polyclonal antibody (Bioss, Woburn, MA, USA) and an HRP-labeled goat anti-rabbit IgG antibody (ZSGB-BIO) for detecting ACROSIN. β-actin was used as a loading control; it was detected using an anti-β-actin monoclonal antibody (ZSGB-BIO) and an HRP-labeled goat anti-mouse IgG antibody (ZSGB-BIO). Proteins were visualized using an Enhanced Chemiluminescence Detection System (Bio-Rad, Hercules, CA, USA). Protein expression on the Western blot was quantitated using Quantity One, a software package from Bio-Rad.
Breeding
Eighteen days after injection of siRNA plasmids, the male mice were placed into cages with cycling females at a male to female ratio of 1:2. Females were checked for vaginal plugs at 0700 h the next morning. The female mouse was considered to be pregnant if a vaginal plug was detected, and it was recorded as 0.5 days post conception (dpc). Then the pregnant female mice were maintained separated from the male and were checked daily for birth. The date of birth and the number of pups were recorded.
Sperm Density, Motility, Testis Weight, Testosterone Level, and Prostate Surface Antigen Staining
Eighteen days after siRNA plasmids injection, male mice were anesthetized with an intraperitoneal injection of 40 mg/kg pentobarbital and their testes and epididymides were surgically removed. Testes were weighed by electronic balance capable of ±0.01 mg accuracy. Sperm were immediately retrieved from the cauda epididymides as described previously (Toshimori et al., 1998). Blood was collected from abdominal aorta and centrifuged at 1500g for 15 min at 4 °C for separating serum. Testosterone level of serum was detected by Cobas e411 automated electrochemiluminescent immunoassay system (Roche, Mannheim, Germany). Blood of male mice 3 days after siRNA plasmids injection was also collected for testosterone detect. Semen volume was determined by direct reading of the graduations on collection tubes. From each semen sample, 100 µL of semen was diluted with isotonic sodium citrate solution at 37 °C (3%, w/v, dissolved in distilled water) at the rate of 1:10. A slide was placed on phase contrast microscope and allowed to warm up to 37 °C; then a small droplet of diluted semen was placed on the slide, and percentage motility was evaluated visually at a magnification of 400x. Motility estimations were performed from 3 different fields in each sample (Bearden and Fuquay, 2000), and the morphology of sperm was observed as well. These evaluations were performed blind to treatments, and the mean of the 3 estimations was used as the final motility score.
The rest spermatozoa were capacitated in modified Krebs-Ringer bicarbonate solution (TYH medium) following methods described elsewhere (Saxena et al., 1999; Toshimori et al., 1998). Aliquots of the spermatozoa (5 × 106 cells/mL) were transferred into a 0.5-mL polypropylene microcentrifuge tube containing 0.2 mL of 4 µg/mL 4-pregnen-3,20-dione in a WB buffer containing 10 mM HEPES, 1.2 mM MgSO4, 5 mM NaHCO3, 5 mM KCl, 130 mM NaCl, 0.15 mM NaH2PO4, 0.7 mM Na2HPO4, 1.5 mM CaCl2, and 5.5 mM D-glucose, pH 7.4) and incubated at 37 °C for 1 h in the dark for acrosome reaction induction. Then, the sperms were fixed in 100% methanol on ice for about 1 h. The cell suspension was centrifuged, and the pellets (about 10 µL) were gently suspended and spotted onto microscope slides. Following air dry, the slides were quickly dipped in acetone, and the sperm smear on the slide was stained with 25 µg/mL FITC-conjugated anti–prostate surface antigen (PSA) antibody (Sigma, St. Louis, MO) in the WB buffer for 10 min in dim light. Control staining with FITC only and inclusion with IgG isotype did not show any staining. The slides were then washed twice in deionized water and fixed in 2% formalin for 15 min. After a thorough rinse in deionized water, the slides were mounted in glycerol and examined under an Olympus immunofluorescence microscope. A minimum of 400 cells/slide were scored for acrosome-intact and partially or totally reacted sperm (Cross et al., 1986) according to the following staining patterns: (1) bright, homogeneous fluorescence in the anterior sperm head region and (2) patchy fluorescence or staining limited to the equatorial segment.
In Vitro Fertilization
Spermatozoa from male mice 18 days after siRNA plasmids injection were also collected for in vitro fertilization (IVF). Capacitation and IVF were performed in TYH medium following methods described elsewhere (Saxena et al., 1999; Toshimori et al., 1998); female mice were given 1 dose of pregnant mare serum gonadotropin (7.5 IU, PMSG; Ningbo Hormone Product Co., Ltd., Ningbo, China), followed 48 h later by human chorionic gonadotropin (hCG; 7.5 IU, Ningbo Hormone Product Co., Ltd.) via intraperitoneal injection to induce superovulation. Oocytes were collected approximately 15 h after hCG injection. Only oocytes in Metaphase II were selected for IVF culturing. Metaphase II oocytes are recognized by the presence of the extruded first polar body (Balakier and Casper, 1991; Huang et al., 1999). Spermatozoa capacitation and IVF were performed in modified Krebs-Ringer bicarbonate solution (TYH medium) following the methods described elsewhere (Saxena et al., 1999; Toshimori et al., 1998). When capacitated sperm were added into the drops of Metaphase II oocyte, the time to disperse the cumulus around the oocytes and to form the male pronucleus in each oocyte was recorded. The number of oocytes and 2-cells embryos was also recorded. The ratio of 2-cell embryos to the total of 2-cell embryos plus metaphase oocytes was recorded as the in vitro fertility rate. These 2-cell embryos were cultured in vitro until the blastocyst stage. The numbers of total blastula and hatched blastula were recorded to calculate the blastula formation rate and blastula hatching rate.
Sperm Enzymatic Assays (Acrosin and Hyaluronidase)
The sperm acrosin activity was determined by a procedure described elsewhere (Kennedy et al., 1989), with modifications. Briefly, for acrosin activity, the following solutions were prepared for this assay: Solution A (11% Ficoll in 0.12 M NaCl, 0.025 M HEPES buffer at pH 7.4), Solution B (0.01% Triton X-100 in 55 mM HEPES, 55 mM NaCl at pH 8.0), Solution C (500 mM benzamidine in water), Solution D (23 mM BAPNA in DMSO), Solution E (1 part Solution B and 3 parts Solution D). Sperm of both groups were adjusted to a concentration of 8 × 106 sperm/mL with PBS, and 0.25 mL of each sperm suspension was placed into a 1.5-mL Eppendorf tube to which 0.5 mL of Solution A was added. After centrifugation at 1500g for 10 min, the supernatant was removed except for 100 µL remaining for the subsequent steps. To the control tube, 100 µL of Solution C was added. Then 1.0 mL of Solution E was added to all tubes, mixed thoroughly by vortexing, and incubated at 23 °C for 3 h; the contents of the tubes were mixed once every hour. After incubation, 100 µL of Solution C was added to end the reaction. Samples were shaken and then centrifuged at 1500g for 10 min. Supernatant (0.2 mL) from both the experimental and control tubes was added to different wells of a 96-well plate and a similar amount of Solution E was added to another well, which served as the zero control. Absorbance was determined using a Microplate Reader at a wavelength of 410 nm. One international unit (IU) of acrosin activity was defined as the amount of enzyme that hydrolyzed 1 mmol BAPNA/min at 23 °C. The following formula was used: Acrosin activity (µIU)/2 × 106 sperms = (ODtest – ODcontrol) × 106/1485 × 2.
Sperm hyaluronidase activity was detected as described previously (Joyce et al., 1986; Wilkinson et al., 1996) with modifications. Sperm samples from mice of both treatment groups were diluted 1 in 5 with 0.15 M sodium chloride to a concentration of about 2 × 107/mL before assay. Aliquots (0.1 mL) of N-acetylglucosamine (NAG) standards (0.5, 2, 4, 10, and 30 µg/mL in water), diluted sperm, and water (reagent blank) were added to the substrate mixture consisted of 0.1 mL acetate buffer (0.3 M containing 0.45 M NaCl, pH 3.8) and 0.1 mL hyaluronic acid substrate (4 mg/mL in water). The mixtures were incubated at 37 °C for 24 h. At the end of the incubation period, the reaction was terminated by heating the mixtures at 100 °C for 5 min; then they were cooled in an ice-water bath. After cooling, samples were centrifuged at 500g for 5 min to remove the spermatozoa. Then 60 µL of potassium tetraborate (0.8 M in water, pH 10.0) was added with mixing before heating for 5 min at 100 °C. The reaction mixtures were cooled in an ice-water bath before adding 2 mL of p-dimethylaminobenzaldehyde (stock p-dimethylaminobenzaldehyde reagent: 10% w/v in 12.5% v/v concentrated hydrochloric acid in glacial acetic acid; stock reagent diluted 1 in 10 with glacial acetic acid before use) and were then incubated for 20 min at 37 °C in a water bath. The reaction mixtures were centrifuged immediately (1500g for 10 min) and the absorbance of the supernatant was read at 582 nm within 30 min. NAG was used as a standard and was reacted with p-dimethylaminobenzaldehyde. One unit of hyaluronidase was defined as the amount that causes the release of 1 µmol equivalent of NAG in 1 h at 37 °C.
Statistical Analysis
IVF rates, blastula formation rates, blastula hatching rates of 2 groups, and the periods of locomotor activity were compared by chi-square test. Other experimental data are presented as mean ± SEM of at least 3 independent experiments. The statistical significance of differences between experimental groups was determined by Student t test (SPSS 11.0, SPSS Inc., Cary, NC). Probability values (p) of less than 0.05 were considered statistically significant.
Results
To determine the role of CLOCK in male fertility, a small hairpin RNA expression plasmid strategy was used to knock down Clock expression. The pClock.shRNA plasmid was introduced into the testes of sexually mature mice. Immunohistochemistry demonstrated reduced CLOCK protein in the testes and the round spermatids at 3 and 6 days after plasmid introduction, relative to testes of mice receiving the control (pCtrl.shRNA) plasmid (Fig. 1A). CLOCK protein was reduced in acrosome of the round spermatids, and this effect was most pronounced at day 3 following plasmid introduction (Fig. 1A, upper panel). Testes of similarly treated mice were harvested and homogenized, followed by Western blotting for CLOCK protein expression. Western blot results confirmed the results of immunohistochemistry, with a significant reduction in CLOCK protein at 3 and 6 days after treatment (p < 0.05; Fig. 1, B and C). These results demonstrate that the shRNA knockdown strategy is effective in knocking down Clock expression in vivo.
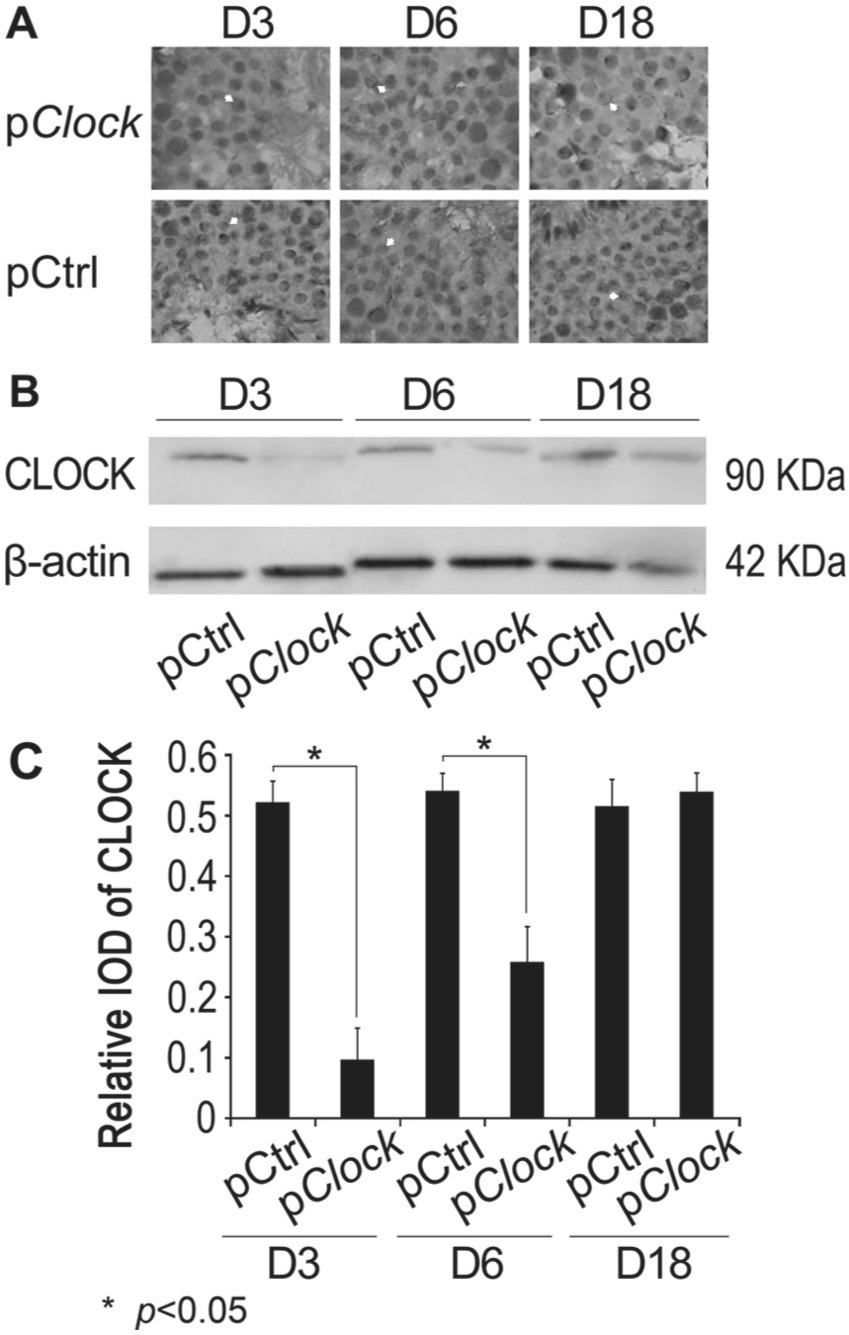
CLOCK knockdown in vivo using a CLOCK shRNA expression plasmid. (A) A Clock shRNA expression plasmid (pClock.shRNA) or its cloning backbone control vector pCtrl.shRNA was transfected into the testes of sexually mature male (n = 9 each group) mice by injection as described in the Materials and Methods. The transfected testes were collected 3, 6, or 18 days post injection for immunohistochemistry staining for CLOCK expression using an anti-CLOCK antibody. The white arrows indicate the stained CLOCK in acrosomes. (B) The testes of transfected mice were harvested and homogenized 3, 6, or 18 days post injection for CLOCK expression by Western blotting using an anti-CLOCK antibody. Western blotting using an anti-β-actin was performed as a loading control. The data are representative of at least 3 independent experiments. (C) Quantitative analysis of data from part B.
We then took advantage of this strategy to determine the effect of CLOCK knockdown on male fertility. First, testes of male mice were transfected with pClock.shRNA, and after 18 days the males were bred with normal female mice of reproductive ages. The 18-day delay between treatment and breeding was designed to allow the gametes generated during the period of Clock gene knockdown to reach maturity. The litter size, determined within a day of birth, was 1.20 ± 1.20 in litters sired by pClock.shRNA transfected males, which was 86.4% lower than the average size of litters sired by the pCtrl.shRNA transfected male mice (8.80 ± 1.46, p < 0.001) (Fig. 2A). However, there was no notable difference in the duration of pregnancy, which was around 19 days in both groups.
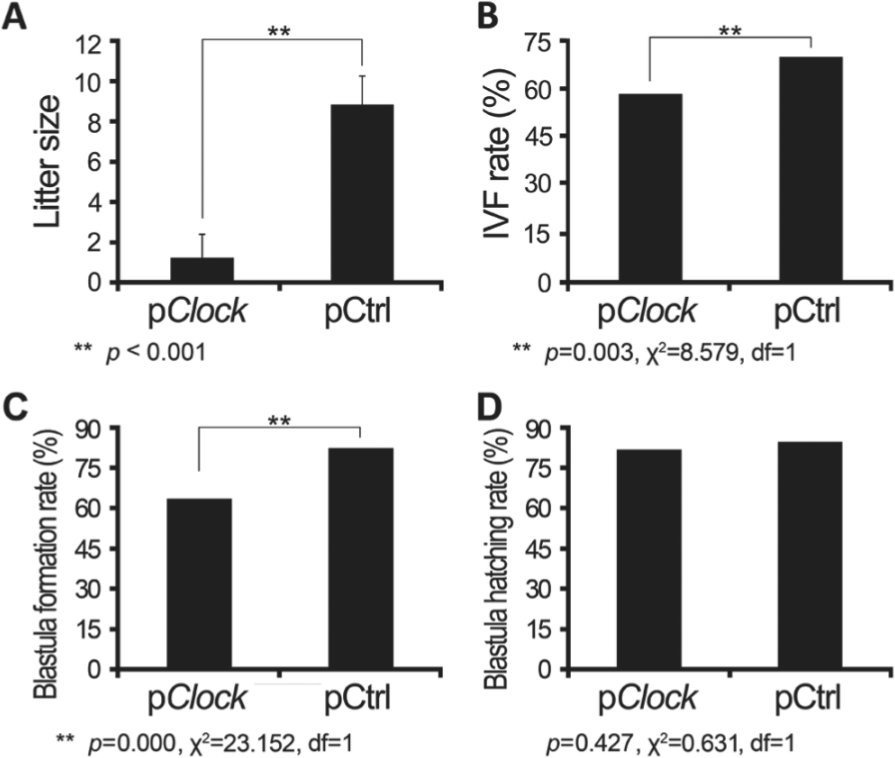
The effect of Clock knockdown on male mouse fertility. The shRNA plasmids (pClock and pCtrl) were transfected into the testes of male mice (n = 20 each group), and 18 days post injection these males were bred with normal female mice. (A) The litter size was determined within 24 hours of birth. Sperms of these shRNA plasmid transfected mice were collected, counted, and compared in terms of their (B) in vitro fertility (IVF), (C) blastula formation rate, and (D) blastula hatching rate. Litter size is presented as mean ± SEM of at least 3 independent experiments, and other data are mean values.
The locomotor activity of the CLOCK-knockdown males was monitored to determine whether their circadian rhythm would be altered by the pClock.shRNA transfection, with comparison to those transfected with pCtrl.shRNA plasmid (Fig. 3, A and B). The mean values of free-running periods in pClock.shRNA group were 23.228 h (before plasmid injection) and 23.416 h (after plasmid injection), and the mean periods in pCtrl.shRNA group were 23.27 h (before plasmid injection) and 23.33 h (after plasmid injection). The results detected little changes in the locomotor activity of these mice (χ2 = 4.368, df = 3, p = 0.224).
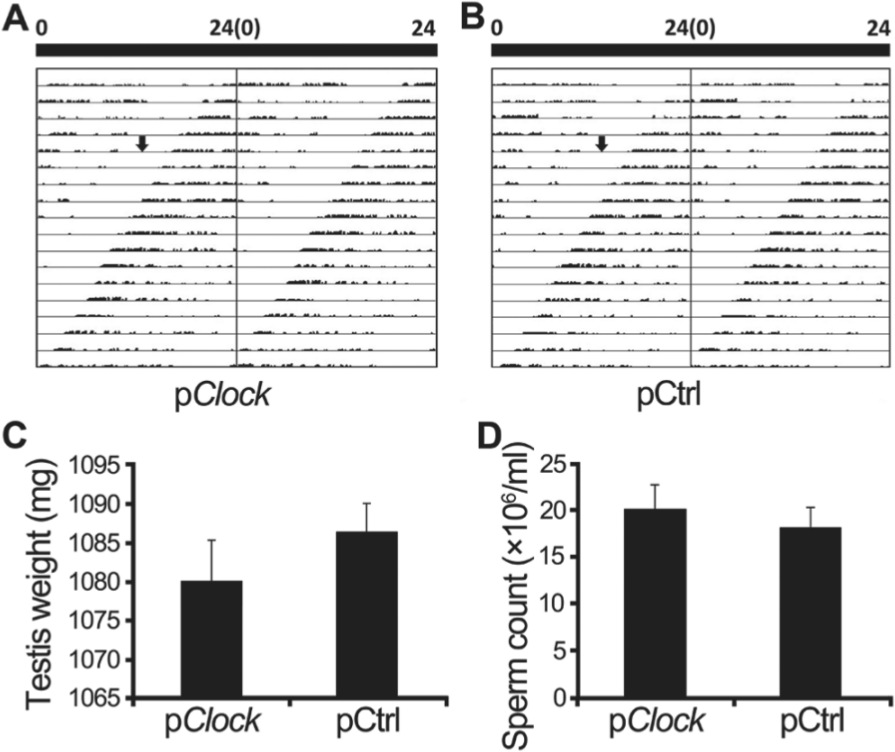
Locomotor activity, testis weight, and sperm count of mice. Male mice were injected with pClock.shRNA, and its cloning backbone control vector pCtrl.shRNA was injected as control. The wheel-running numbers were recorded to present locomotor activity, which was presented as a double-plotted actogram (A and B). On the double-plotted actogram, the arrow indicates the injection time. The testes of these shRNA plasmid transfected mice were collected 18 days post injection, their weights were assessed (C), and sperm were collected and counted (D). Data are presented as mean ± SEM of at least 3 independent experiments.
To investigate whether the smaller litter size of the CLOCK-knockdown male mice was attributable to the changes in the testes function or sperm development and function, testes, blood serum and sperm were collected and compared. The testis weights of the 2 groups did not differ significantly (p = 0.424, Fig. 3C), nor did the testosterone levels in blood serum of male mice 3 or 18 days post plasmid injection (p = 0.401 at day 3, p = 0.379 at day 18, Fig. 4D). The testosterone level of pCtrl.shRNA transfected mice was 6.273 ± 0.461 ng/mL at day 3 and 6.017 ± 0.532 ng/mL at day 18 and that of pClock.shRNA transfected mice was 5.637 ± 0.575 ng/mL at day 3 and 6.084 ± 0.596 ng/mL at day 18. The sperm counts—(18.20 ± 2.18) × 106/mL for the pCtrl.shRNA transfected group and (20.12 ± 2.66) × 106/mL for the pClock.shRNA transfected group—also did not differ significantly (p = 0.168, Fig. 3D). In both groups, all sperm samples had moderate to strong motility.
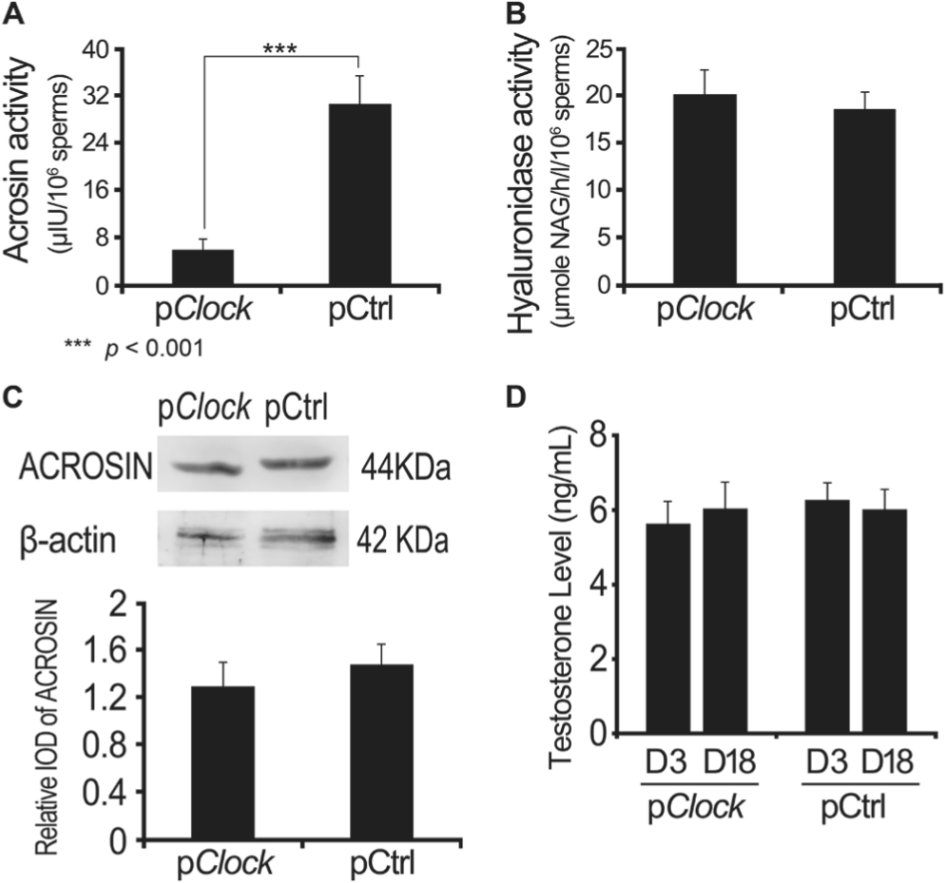
The effect of Clock knockdown on acrosomal enzymes and testosterone. At 18 days post injection of shRNA plasmids (pClock and pCtrl), the male mice were anesthetized and their cauda epididymis sperm and blood were collected. The activities of acrosin (A) and hyaluronidase (B) were detected, as was the ACROSIN level (C). The serous testosterone level was also measured (D). Data are mean ± SEM of at least 3 independent experiments.
The morphology of sperms and their acrosomes were compared by immunofluorescence staining using an antibody against PSA (Fig. 5A). The percentage of acrosome-intact spermatozoa (AIS%) was found to be 87.56 ± 0.58 for the pCtrl.shRNA transfected group and 88.18 ± 0.49 for the pClock.shRNA transfected group (Fig. 5B), and the difference was insignificant (p = 0.438). These results suggest that CLOCK knockdown does not affect testicular weight, testosterone level, or sperm production.
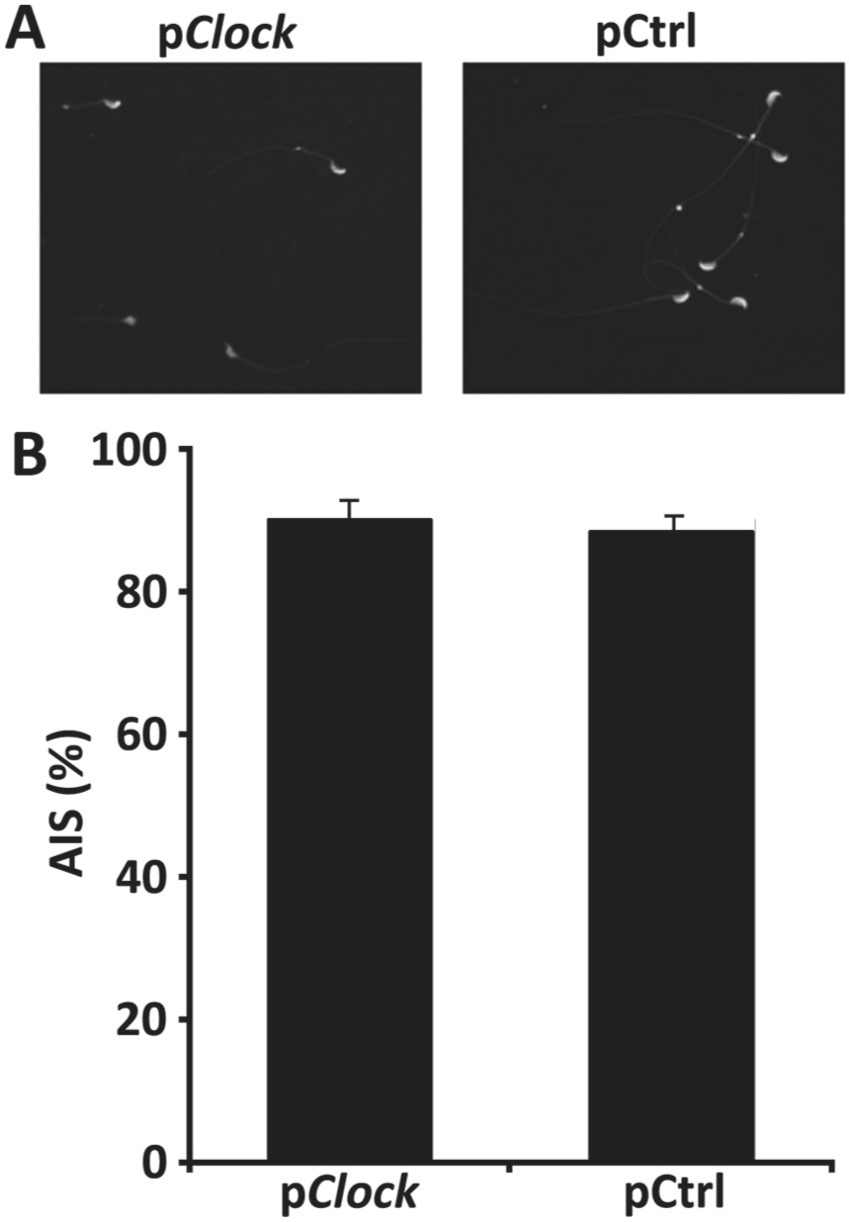
Detection of the influence of Clock knockdown on sperm formation. At 18 days post transfection with the shRNA plasmids (pClock and pCtrl), the sperms of these males were collected and subjected to immunofluorescence staining using an anti-PSA antibody (A). Acrosome-intact spermatozoa (AIS) were counted and expressed as a percentage of the total number of sperms (AIS%) (B).
We further characterized the functionality of sperm from mice treated with pClock.shRNA and pCtrl.shRNA by assessing in vitro fertilization, sperm enzymatic activities, and ACROSIN level. Just 5 min after sperm was added to the oocyte drops, the cumulus was totally dispersed from the oocytes of pCtrl.shRNA group; however, from the oocytes of pClock.shRNA group, the cumulus was observed to disperse on the 7th min after sperm addition and was totally dispersed on the 15th to 16th min. During in vitro fertilization, this slight delay of about 10 min was observed only in the sperm of pClock.shRNA transfected mice dispersing cumulus cells. Ova were collected from 20 female mice for in vitro fertilization with sperm from each of the 2 male treatment groups. The average number of ova per female (29.9 vs. 25.2) and the percentage of ova classified as being in metaphase II before addition of sperm (468 of 598, or 78.3%, and 384 of 503, or 76.3%) were comparable in the groups used for IVF with the pClock.shRNA and pCtrl.shRNA sperm, respectively. In contrast, the success of IVF differed significantly between the groups, as assessed by the percentage of ova in Metaphase II that progressed to the 2-cell stage. 275/468 and 263/384, for pClock.shRNA and pCtrl.shRNA groups, gave IVF rates of 58.76% and 68.49%, respectively (Fig. 2B) (χ2 = 8.579, df = 1, p = 0.003). The blastula formation rate was calculated as the percentage of 2-cell embryos that progressed to the blastocyst stage and the percentage of blastula that hatched out from the zona pellucida recorded as the blastula hatching rate. The blastula formation rate was 63.6% (175/275) for pClock group and 82.1% (216/263) for pCtrl group; these 2 rates were significantly different (Fig. 2C) (χ2 = 23.152, df = 1, p = 0.000). However, the blastula hatching rate was of no significance, at 81.7% for pClock group and 84.7% for pCtrl group (Fig. 2D) (χ2 = 0.631, df = 1, p = 0.427).
The acrosin activity was 30.69 ± 4.76 µIU/106 sperms for the pCtrl.shRNA group and 5.94 ± 1.85 µIU/106 sperms for the pClock.shRNA group (Fig. 4A), a highly significant difference (p < 0.001). However, the hyaluronidase activity did not differ significantly between the sperm from the 2 groups (Fig. 4B, p = 0.54). Taken together, these results demonstrated that CLOCK knockdown in male mice led to smaller litter size, a lower IVF rate, and lower acrosin activity of the sperm. Proteins of similarly treated mice sperm were harvested and homogenized, followed by Western blotting for ACROSIN protein expression. Western blot results showed a slight reduction in ACROSIN protein without significance (p > 0.05; Fig. 4C). These results demonstrate that CLOCK knockdown decreases the activity of acrosin but not its expression level.
Discussion
In this study, we used the small hairpin RNA strategy to knock down CLOCK expression in mouse testes and round spermatid, finding that CLOCK knockdown led to a significant decrease in the offspring produced by these male mice (Fig. 2). To help identify the possible role of the Clock gene in the round spermatid and testes, locomotor activity of the mice was monitored, and testicular weight, testosterone level, sperm production, and functionality were systemically examined. No difference was observed in locomotor activity or acrosin expression level, and no apparent abnormalities were found in testicular weight, testosterone level, or sperm production. The slight decrease of testes weight might be due to the mild decline in testosterone level, which is essential for spermatogenesis (Walker, 2011), but no significance was observed in either testes weight or testosterone level. A lower IVF rate, a lower blastula formation rate, and a lower acrosin activity were found in the CLOCK knockdown sperms, as well as a delay in dispersing cumulus cells. These results demonstrate that acrosin activity could be regulated by CLOCK and that CLOCK contributes to the regulation of male fertility and blastula formation. These findings not only provide new insights into the functions of the Clock gene but may also lead to development of novel contraceptive strategies.
Following CLOCK knockdown, the greatest reduction in CLOCK immunostaining was observed in round spermatids 3 days following the pClock.shRNA plasmid transfection (Fig. 1). Since the haploid round spermatids require 2 weeks to differentiate into the species-specific shaped spermatozoon (Clermont and Trott, 1969; Hecht, 1998), we chose to breed the animals or collect sperm from the animals for analysis 18 days after the shRNA plasmid transfection.
Our results showed that there was a lower IFV rate and a lower blastula formation rate when CLOCK knockdown sperm were used (Fig. 2, B and C), but no difference was observed in the blastula hatching rate (Fig. 2D). As the difference in the IVF rate was not as great as the decrease in litter size, it appears that the decreased fusion of sperm with oocytes and an influence on early embryonic development together led to the small litter size. During spermatogenesis, CLOCK expression is restricted to round spermatids and the developing acrosome (Alvarez et al., 2003). We assessed activities of 2 of the main acrosomal enzymes and found that acrosin activity of Clock knockdown sperm was significantly decreased (Fig. 4A), which was not induced by its expression reduction (Fig. 4C), while hyaluronidase activity was not (Fig. 4B). Mice with sperm lacking the acrosin protease activity exhibit a delay in penetration of the zona pellucida, suggesting that acrosin plays a major role in acceleration of the dispersal of acrosomal proteins (Yamagata et al., 1998). The lower acrosin activity in CLOCK knockdown sperm may account for the 10-min delay of CLOCK knockdown sperm in dispersing cumulus cells.
Previous studies of the role of Clock in reproductive function have described a modest decrease in reproductive performance in mice homozygous for a dominant-negative mutant allele, ClockΔ19/Δ19 (Dolatshad et al., 2006). In view of the marked impact of shRNA-mediated knockdown of CLOCK, it seems a bit surprising that the phenotype of the ClockΔ19/Δ19 mutant mice is not more severe. Even more surprising is the maintenance of reproductive performance of male mice homozygous for a null allele of Clock. This apparent discrepancy with the marked effects of transient gene disruption reported here suggests that the long-term absence of functional CLOCK protein may lead to compensatory upregulation of a related gene, buffering the long-term impact of CLOCK deficiency. The reduction in fertility observed following Clock.shRNA may reflect the critical importance of these “circadian transcription factor complexes” in a vital noncircadian role.
In summary, we demonstrated that expression of the circadian gene, Clock, can be effectively attenuated in testes of mice by RNAi targeting Clock without altering their rhythm of locomotor activities. We found that CLOCK knockdown leads to a reduced fertility (revealed by the lower IVF rate, lower blastula formation rate, and smaller litter size of the CLOCK knockdown male mice) and down-regulation of acrosin activity in the sperm, which might be responsible for the delayed dispersion of cumulus cells. These results provide the first evidence to support an essential role of the circadian gene, Clock, in male fertility. Regulation of the acrosome activity by Clock gene warrants further investigation.
Footnotes
Acknowledgements
The authors wish to acknowledge membership within and support from the National Nature Science Foundation of China (No. 41074131 to ZW), China Medical Board (CMB) (No. 88-486 to ZW), and Sichuan University “985 project—Science and technology innovation platform for novel drug development.” We thank David Weaver (UMass Medical School, Worcester, MA, USA) and Johnny He (UNT Health Science Center, Fort Worth, TX, USA) for comments and for editing the manuscript.
Conflict of Interest Statement
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.