
Abstract
Aim
This study aimed to evaluate the efficacy and tolerability of rimegepant for the prevention of episodic migraine in participants with a documented history of inadequate response to 2–4 categories of traditional oral preventive medication (OPM).
Methods
This multinational phase 4 trial consisted of an untreated 28-day observational phase (OP) and a 12-week double-blind treatment (DBT) phase. Participants with 4–14 monthly migraine days (MMDs), <15 monthly headache days (<7 non-migraine) and documented previous inadequate response to 2–4 traditional OPM categories were enrolled. Participants were randomized to rimegepant 75 mg orally disintegrating tablet (ODT) or placebo every other day (EOD). The primary endpoint was mean change from the OP in MMDs through the 12-week DBT phase. Key secondary endpoints were tested hierarchically to control type I errors. Tolerance and safety were assessed throughout the DBT phase.
Results
In total, 328 participants received rimegepant and 324 received placebo. The most common OPM categories with prior inadequate response were anticonvulsants (61%), beta-blockers (56%) and amitriptyline (51%). The mean ± SD number of MMDs in the OP was 8.4 ± 2.4 and 8.3 ± 2.3, respectively, in the rimegepant (n = 324) and placebo (n = 319) groups. Across the DBT phase, participants who received rimegepant had a significantly larger mean change from the OP in MMDs than those who received placebo (−2.1 vs. −0.5 days; difference = −1.6 days; 95% confidence interval (CI) = −2.1 to −1.2; p < 0.0001). All key secondary endpoints favored rimegepant: (i) percentage of participants with ≥50% reduction from the OP in MMDs with moderate or severe pain intensity across the DBT phase (difference: 20.1%; 95% CI = 13.7 to 26.5; p < 0.0001); (ii) mean change from the OP in MMDs in the first month of the DBT phase (difference: −1.7 days; 95% CI = −2.3 to −1.2; p < 0.0001); (iii) mean change from the OP in MMDs in the last month of the DBT phase (difference: −1.4 days; 95% CI = −2.1 to −0.8; p < 0.0001); (iv) mean change from baseline in Migraine-Specific Quality-of-Life Questionnaire v2.1 Restrictive Role Function domain score at week 12 of the DBT phase (difference: 6.6 points; 95% CI = 3.6 to 9.5; p < 0.0001); and (v) mean change from baseline in Migraine Interictal Burden Scale score at week 12 of the DBT phase (difference: −0.9 points; 95% CI = −1.4 to −0.4; p = 0.0006). Rimegepant was well tolerated with a safety profile not notably different from placebo.
Conclusions
Rimegepant 75 mg ODT EOD is efficacious and well tolerated for the prevention of episodic migraine in participants with a documented history of inadequate response to 2–4 categories of traditional OPM.
Trial Registration
ClinicalTrials.gov, NCT05518123 (https://clinicaltrials.gov/study/NCT05518123).
This is a visual representation of the abstract.
Introduction
Migraine affects approximately one in seven people worldwide and is responsible for approximately 5% of global ill-health (1). As a neurologic disease, migraine is the second largest cause of disability in the global population and the most common cause of disability among young women (2). Although not life-threatening, migraine is a significant personal burden on those affected, both during attacks and between them (3–5). Since migraine prevalence is highest among people of working age, this also translates into a notable societal burden, particularly in terms of lower productivity and higher healthcare utilization (1,3–5).
Effective relief from the symptoms of migraine attack is a mandatory clinical aim; however, the ideal outcome would be the prevention of attacks altogether. Improved migraine control with preventive treatment is directly linked to a reduction in the frequency and severity of attacks, which allows people living with migraine to build a better quality of life and reduce their mental health burden (6–9). Traditional oral preventive medications (OPMs), such as beta-blockers, anticonvulsants, and tricyclic antidepressants, were not specifically designed to treat migraine (7). Adherence to traditional OPMs can be poor, often due to lack of adequate efficacy or intolerance (7,8,10–14). For many, this is compounded by a need to titrate towards a therapeutic dose, which can delay efficacy by several months (7,8,10–14). These factors have led to an unmet need for alternative OPMs that is particularly felt among those who have tried and failed to obtain adequate clinical benefit from multiple previous therapies (15,16).
Due to their favorable efficacy and safety profiles, therapies that target the neuropeptide calcitonin gene-related peptide (CGRP) are now recommended among the first-line therapies for the prevention of episodic migraine and represent alternatives to traditional OPMs (11,14,17,18). In many regions of the world, monoclonal antibodies against the CGRP peptide or its receptor have been approved for prophylactic use; however, these are dosed either intravenously or subcutaneously, and research suggests that people with migraine prefer oral medications (19–23). Two small-molecule antagonists of the CGRP receptor, namely rimegepant and atogepant, are oral medications approved for preventive therapy, with rimegepant also approved for the acute treatment of migraine attacks (24–27). Two clinical trials have shown rimegepant 75 mg to significantly reduce the frequency of episodic migraine attacks when taken every other day (EOD) (28–30). Both trials showed a significantly larger decrease in the mean monthly migraine days (MMDs) with rimegepant versus placebo, with efficacy observed within the first month of treatment and a safety profile not notably different than placebo (28–30).
This phase 4 trial aimed to be the first to rigorously evaluate the efficacy and tolerability of rimegepant for the prevention of episodic migraine in participants with documented previous inadequate response to 2–4 categories of traditional OPM in the previous 10 years. Inadequate response was defined as a lack of efficacy, prior intolerance, or contraindication. At least one of the documented categories with inadequate response must have been due to lack of efficacy or prior intolerance.
Methods
Study design
A multinational, phase 4, randomized, double-blind, placebo-controlled trial (NCT05518123; BHV3000-407) was designed in compliance with the International Headache Society Guidelines for controlled trials of preventive treatment in adults with episodic migraine (31). The trial consisted of a 28-day observation phase (OP), a 12-week double-blind treatment (DBT) phase, a 12-week open-label extension (OLE) phase and a two-week safety follow-up phase (Figure 1). Participation in the OLE phase was optional and is ongoing at the time of writing. This report considered data from the OP and DBT phases only. In-person study visits occurred at screening, at the end of the OP (pre-randomization visit, within four days of baseline), at the start of the DBT phase (baseline), at weeks 2, 4, 8 and 12 of the DBT phase (or at the end of treatment if discontinuing before week 12 of the DBT phase), and at the safety follow-up for those who did not continue to the OLE phase (within 14 days of the week 12/end of treatment visit).

Study design. BLV = baseline visit; DBT = double-blind treatment; eDiary = electronic diary; EOD = every other day; MIBS = Migraine Interictal Burden Scale; MSQ = Migraine-Specific Quality of Life Questionnaire V2.1; ODT = orally disintegrating tablet; OLE = open-label extension; OP = observation phase; wks = weeks.
Ethical statement
The trial was conducted in compliance with the Declaration of Helsinki, International Conference on Harmonisation guidelines, Good Clinical Practice, Good Laboratory Practice and all locally applicable regulations. All participants provided their written informed consent. The protocol and consent forms were approved by the Institutional Review Board/Independent Ethics Committee at each site prior to initiation of the study (see supplementary material, Table S1).
Participants
To enroll, participants were required to be at least 18 years of age with migraine onset prior to 50 years of age and at least a one-year history of migraine attacks (with/without aura) per the International Classification of Headache Disorders, 3rd edition (ICHD-3) criteria (32). Other key eligibility criteria included untreated migraine attacks lasting an average of 4–72 hours each; an average of 4–14 migraine days per month (considered 28 days for practical purposes) over the three months prior to screening and during the OP; an average of fewer than 15 headache days (migraine or non-migraine) per month over the three months prior to screening and during the OP; an average of fewer than seven non-migraine headache days per month over the three months prior to screening and during the OP; and documented previous inadequate response (due to lack of efficacy, prior intolerance or contraindication) to 2–4 traditional OPM categories.
A migraine day was defined as a day with a headache lasting at least 30 minutes and either (i) nausea/vomiting, or photophobia and phonophobia or (ii) two or more of the following headache features: unilateral pain, pulsating pain, moderate or severe intensity pain, or pain aggravated by/causing avoidance of physical activity. Days during the trial on which acute migraine-specific medication (i.e. triptan, ergotamine (although not permitted), lasmiditan or ubrogepant) was taken were also classified as migraine days, regardless of pain intensity or symptoms. MMDs were calculated based on a 28-day month. A headache day was defined as either (i) a migraine day (as above); (ii) a day with a non-migraine headache lasting at least 30 minutes; or (iii) a day on which medication was taken to treat a headache or aura.
Prior inadequate response was defined as lack of efficacy in the previous 10 years, prior intolerance in the previous 10 years or presence of a contraindication. At least one qualifying incidence of inadequate response to a prior OPM must have been due to lack of efficacy or prior intolerance. Lack of efficacy was defined as no meaningful reduction in headache frequency or severity (i.e. less than 50%) after administration for a clinically adequate period of time at the accepted therapeutic dose. Intolerance was defined as interruption and discontinuation of a medication due to an adverse event (AE). Contraindications were per labeling, practice guidelines, or current standard of care. Each qualifying instance of inadequate response required documentation of the participant's OPM use and reason for discontinuation/rationale for inadequate response in their medical/pharmacy records. If records were incomplete, OPM use and rationale for claiming inadequate response could be verified through a detailed and documented interview with the treating physician (where the medical/pharmacy records were not available) or participant (where medical/pharmacy records were available but documentation describing the reason for treatment failure was inadequate). OPM categories were: (i) valproic acid; (ii) other anticonvulsants, specifically gabapentin or topiramate; (iii) beta-blockers, specifically atenolol, bisoprolol, metoprolol, nadolol, propranolol or timolol; (iv) tricyclic antidepressant, specifically amitriptyline; (v) serotonin-norepinephrine reuptake inhibitors, specifically desvenlafaxine or venlafaxine; (vi) calcium channel blocker, specifically flunarizine or verapamil; (vii) angiotensin blocker, specifically, candesartan or lisinopril; and (viii) other locally approved or recognized preventive medication.
Participants were excluded if they had a history of cluster headache, migraine with brainstem aura, or hemiplegic migraine; current medication overuse headaches; currently uncontrolled, unstable or recently diagnosed cardiovascular disease (within six months); or other pain syndromes that might interfere with study assessments. Use of preventive medications within the 30 days prior to screening (12 weeks for botulinum injection and 24 weeks for injectable monoclonal CGRP antibodies) and through the study was not permitted. Non-narcotic analgesics should not have been used for more than 14 days per month and narcotics for more than three days per month during the three months prior to screening. Stable use of preventive medications for non-migraine indications was permitted (e.g. beta-blocker to treat a cardiovascular comorbidity).
Intervention
Eligible participants were stratified into four groups by the number of recorded migraine days in the first 28 days of the OP (4–7 or 8–14) and the number of OPM categories with prior inadequate response (<3 or ≥3), then randomized 1:1 to rimegepant 75 mg or placebo orally disintegrating tablet (ODT) via an interactive web response system. Rimegepant and placebo study medications looked and tasted identical, and all persons involved in the trial were blinded to treatment identity until after the database lock. Participants were instructed to administer study medication by placing it on top or under the tongue until fully dissolved, then swallow. Study medication was instructed to be taken EOD throughout the DBT phase, irrespective of migraine symptoms. The time of dosing was to be consistent throughout the study (e.g. first thing in the morning EOD).
Study medication could not be used as treatment for an acute migraine attack in any phase. Permitted standard of care treatments for acute migraine were: non-steroidal anti-inflammatory drugs, acetaminophen (up to 2 g/day for a maximum of two consecutive days), triptans, antiemetics, baclofen and other locally approved medications (e.g. lasmiditan or ubrogepant). Narcotics and ergotamine were prohibited acute migraine medications. Their use, if any, was recorded as a protocol deviation.
Measures
Migraine characteristics (pain, features and symptoms) and acute headache medication use were recorded throughout the OP and DBT phase in a daily electronic diary (eDiary). All medication use was also recorded in a daily paper diary. Participants with less than 24 days of completed eDiary reports in the OP were discontinued from the study. At baseline and weeks 4, 8 and 12 of the DBT phase, participants self-completed the Migraine-Specific, Quality-of-Life Questionnaire v2.1 (MSQ) (33) and the Migraine Interictal Burden Scale (MIBS) (15). AEs during the DBT phase were recorded. Clinical safety laboratory testing was conducted within four days of baseline and at week 12 of the DBT phase. Liver function tests were conducted within four days of baseline and at weeks 4 and 12 of the DBT phase.
Endpoints
The primary endpoint was the mean change from the OP in MMDs across the DBT phase (i.e. weeks 1–12). A similar primary endpoint has been used to evaluate migraine prophylaxis in other completed or ongoing trials (34–38).
Key secondary endpoints included: (i) percentage of participants with at least a 50% reduction from the OP in MMDs with moderate or severe pain intensity across the DBT phase; (ii) mean change from the OP in MMDs in the first month of the DBT phase (i.e. weeks 1–4); (iii) mean change from the OP in MMDs in the last month of the DBT phase (i.e. weeks 9–12); (iv) mean change from baseline in MSQ Restrictive Role Function domain score at week 12 of the DBT phase; and (v) mean change from baseline in MIBS score at week 12 of the DBT phase.
Tolerance and safety were assessed during the DBT phase via AE reporting and laboratory/liver function testing. An AE was defined as any new untoward medical occurrence or worsening of a pre-existing medical condition.
Statistical analysis
Based on findings from previous trials, it was estimated that rimegepant could prevent approximately one migraine day per 28 ± 3.6 days (mean ± SD) more than placebo (28,39–41). Approximately 600 participants were planned to be randomized (300 per treatment group) to have approximately 90% power to evaluate the primary outcome at a two-sided alpha level of 0.05.
The primary endpoint was assessed in the migraine analysis set; defined as randomized participants who took at least one dose of DBT medication and had at least 14 days of eDiary data in both the OP and at least one month (28 days) in the DBT phase. Least squares mean change were calculated using a linear mixed-effect model with repeated measures that included change from the OP in MMDs as the dependent variable, and fixed effects of treatment group, randomization stratum, visit (i.e., week 4, 8, or 12), and visit-by-treatment group interaction.
Each key secondary endpoint was tested hierarchically in the numerical order listed above at a two-sided alpha level of 0.05 to control type 1 error. All key secondary endpoints were assessed in the migraine analysis set, except for mean change in MSQ Restrictive Role Function score and MIBS, which were assessed in the evaluable efficacy analysis set, defined as randomized participants who took at least one dose of DBT medication and had non-missing scores at both baseline and at least one scheduled visit at week 4, 8 or 12 of the DBT phase. The percentage of participants with at least a 50% reduction from the OP in MMDs with moderate or severe pain intensity was assessed using Mantel–Haenszel risk estimation stratified by randomization stratum, where missing data were imputed as failure. Key secondary endpoints related to change in MMDs in the first or last month were based on the same model as the primary endpoint. Change in MSQ Restrictive Role Function and MIBS scores were assessed with least squares mean change using a linear mixed-effects model with repeated measures that included change from baseline as the dependent variable; baseline score as a covariate; and fixed effects for treatment group, randomization stratum, visit (i.e. week 4, 8 or 12) and visit-by-treatment group interaction.
Safety and tolerability were assessed descriptively for the safety analysis set, defined as participants who took at least one dose of DBT medication. Adverse events of scientific and regulatory interest were identified before database unblinding. The incidence of medication overuse headache, Raynaud's phenomenon and constipation were each summarized using Medical Dictionary for Regulatory Activities (MedDRA) preferred terms. Hepatic-related AEs included all preferred terms in the hepatic disorders standardized MedDRA Query (SMQ), except for those in the congenital, familial, neonatal, and genetic disorders of the liver SMQ. Potential drug abuse AEs included all preferred terms in the drug abuse, dependence and withdrawal SMQ, plus additional selected terms from the US Food and Drug Administration Guidance for Industry Assessment of Abuse Potential of Drugs (42), and where a preferred term of dizziness was reported concurrent with a euphoria-related preferred term. Cardiovascular AEs included narrow terms from the central nervous system hemorrhages and cerebrovascular accidents, ischemic central nervous system vascular conditions, ischemic colitis, myocardial infarction and other ischemic heart disease SMQs, and all from the embolic and thrombotic events, arterial SMQ. Suicidality AEs included all terms from the suicide/self-injury SMQ. Hypertension AEs included narrow terms from the hypertension SMQ.
Laboratory test results were graded as 0–4 using numerical laboratory test criteria from the Common Terminology Criteria for Adverse Events, version 5, if available, otherwise according to the Division of Acquired Immune Deficiency Syndrome Table for Grading the Severity of Adult and Pediatric Adverse Events Corrected, version 2.1.
Results
Participants
Between November 2022, and December 2024, 962 participants were enrolled in the study (Figure 2). Of these, 652 (rimegepant ODT 75 mg, n = 328; placebo, n = 324) were randomized to study treatment and took at least one dose. These participants were included in the safety set. The majority of participants were in Europe (n = 554, across 10 countries: Austria, Belgium, Denmark, Finland, Germany, Italy, Poland, Spain, Sweden and UK); others were in North America (n = 92, across three countries: Canada, Mexico and USA) or Australia (n = 6). Nine participants did not provide the minimum amount of eDiary data to be included in the migraine analysis set for assessment of the primary endpoint. In all, 624 participants completed the DBT phase; the most common reasons for discontinuation were withdrawal of consent (n = 15) and AE (n = 8). In total, 602 participants continued to the OLE phase, which is ongoing.

Participant disposition. One participant completed the DBT phase but did not continue to the safety follow-up or OLE phase. DBT = double-blind treatment; OLE = open-label extension; OP = observation phase.
Key demographics and baseline clinical characteristics were similar between rimegepant and placebo groups in the safety analysis set (Tables 1 and 2). The mean age among all participants was 42 years and 86% were female. Mean body mass index was 25 kg/m2. Overall, 77% of participants primarily experienced migraine attacks without aura and the historical most bothersome symptom was photophobia in 46% of participants, nausea in 41% and phonophobia in 12%. In the migraine analysis set, mean MMDs during the OP was 8.4 with any pain intensity and 7.1 with moderate or severe pain intensity.
Demographics and clinical characteristics.

*Migraine analysis set. BMI = body mass index; MIBS = Migraine Interictal Burden Scale; MMD = monthly migraine day; MSQ = Migraine-Specific Quality of Life Questionnaire V2.1; OP = observation phase; SD = standard deviation.
Previous inadequate response to traditional oral preventive medicines for episodic migraine.

*Nortriptyline (n = 9), zonisamide (n = 9), nebivolol (n = 6), magnesium (n = 4), pizotifen (n = 4), nortriptyline hydrochloride (n = 3), riboflavin (n = 3), cinnarizine (n = 1), lamotrigine (n = 1) and pregabalin (n = 1).
Participants may have had recorded inadequate response to more than one medication in each category.
Data are n (%). CCB = calcium channel blocker; OPM = oral preventive medication; SNRI = serotonin-norepinephrine reuptake inhibitor; TCA = tricyclic antidepressant.
Approximately one-third (35%) of all participants had documented previous inadequate response to at least three traditional OPM categories. The most common OPM categories with previous inadequate response were anticonvulsants (61%; including 58% topiramate and 4% gabapentin (some participants had failed both)), beta-blockers (56%) and tricyclic antidepressants (i.e. amitriptyline; 51%). No participants reported prior inadequate response to a CGRP-targeting therapy. Reasons for previous inadequate response to at least one OPM category were lack of efficacy in 92% of participants, prior intolerance in 46% and contraindication in 5%.
Mean ± SD time on study medication was 12.0 ± 1.8 weeks in the rimegepant group and 11.9 ± 1.8 weeks in the placebo group. The mean ± SD exposure per month (28 days) was 14.0 ± 1.2 ODTs in the rimegepant group and 14.0 ± 1.0 ODTs in the placebo group. These findings indicate a very high compliance with EOD dosing.
Primary endpoint
Across the DBT phase, participants in the rimegepant group had a mean change of −2.1 (95% confidence interval (CI) = −2.5 to −1.7) MMDs from the OP, whereas participants in the placebo group had a change of −0.5 (−0.8 to −0.1). Therefore, participants who took rimegepant had a 1.6-day greater reduction in MMDs than those who took placebo (95% CI = −2.1 to −1.2; p < 0.0001) (Figure 3).

Primary endpoint: mean change from the observation phase in monthly migraine days across the double-blind treatment phase. CI = confidence interval; DBT = double-blind treatment; LS = least-squares; MMD = monthly migraine day; OP = observation phase; SD = standard deviation.
Key secondary efficacy endpoints
All five key secondary efficacy endpoints were statistically significant following hierarchical testing, demonstrating the statistical superiority of rimegepant versus placebo (Figure 4). For the first key secondary efficacy endpoint, a greater percentage of participants who took rimegepant versus placebo achieved at least a 50% reduction from the OP in MMDs with moderate or severe pain intensity (difference: 20.1%; 95% CI = 13.7 to 26.5; p < 0.0001) (Figure 4A). For the second endpoint, participants who took rimegepant had a 1.7-day greater mean reduction from the OP in MMDs during the first month of the DBT phase (95% CI = −2.3 to −1.2; p < 0.0001) (Figure 4B). For the third endpoint, participants who took rimegepant had a 1.4-day greater mean reduction from the OP in MMDs during the last month of the DBT phase (95% CI = −2.1 to −0.8; p < 0.0001) (Figure 4C). For the fourth endpoint, participants who took rimegepant had a 6.6-point (95% CI = 3.6 to 9.5; p < 0.0001) greater mean increase from baseline in MSQ Restrictive Role Function domain score at week 12 of the DBT phase (Figure 4D). For the fifth endpoint, participants who took rimegepant had a 0.9-point (95% CI = −1.4 to −0.4; p = 0.0006) greater mean reduction from baseline in MIBS score at week 12 of the DBT phase (Figure 4E).
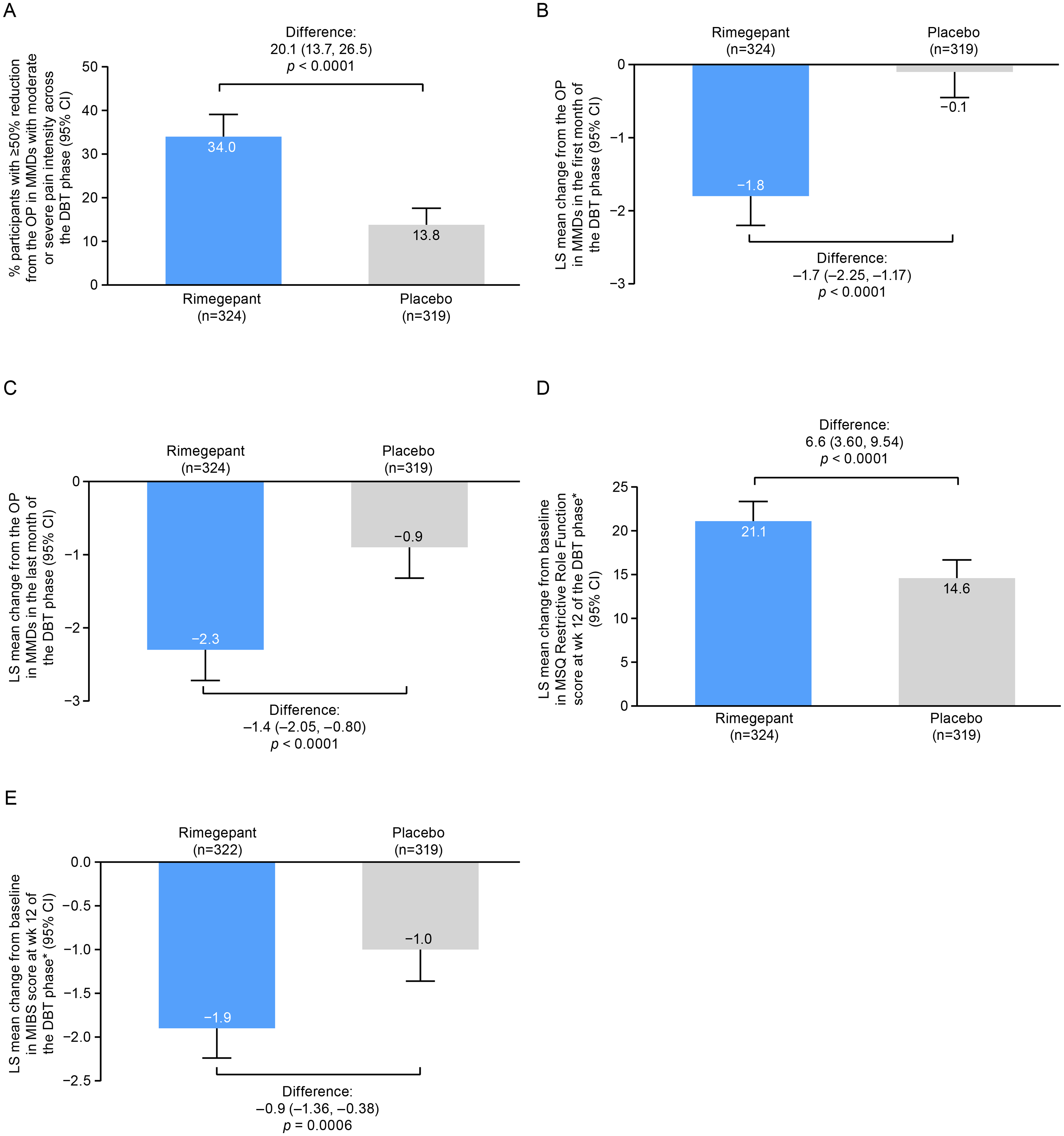
Key secondary endpoints. *All treated participants with non-missing scores at both baseline and ≥1 scheduled visit (week 4, 8 or 12) in the DBT phase. CI = confidence interval; DBT = double-blind treatment; LS = least-squares; MIBS = Migraine Interictal Burden Scale ; MMD = monthly migraine day; MSQ = Migraine-Specific Quality of Life Questionnaire V2.1; OP = observation phase; wk = week.
Tolerability and safety
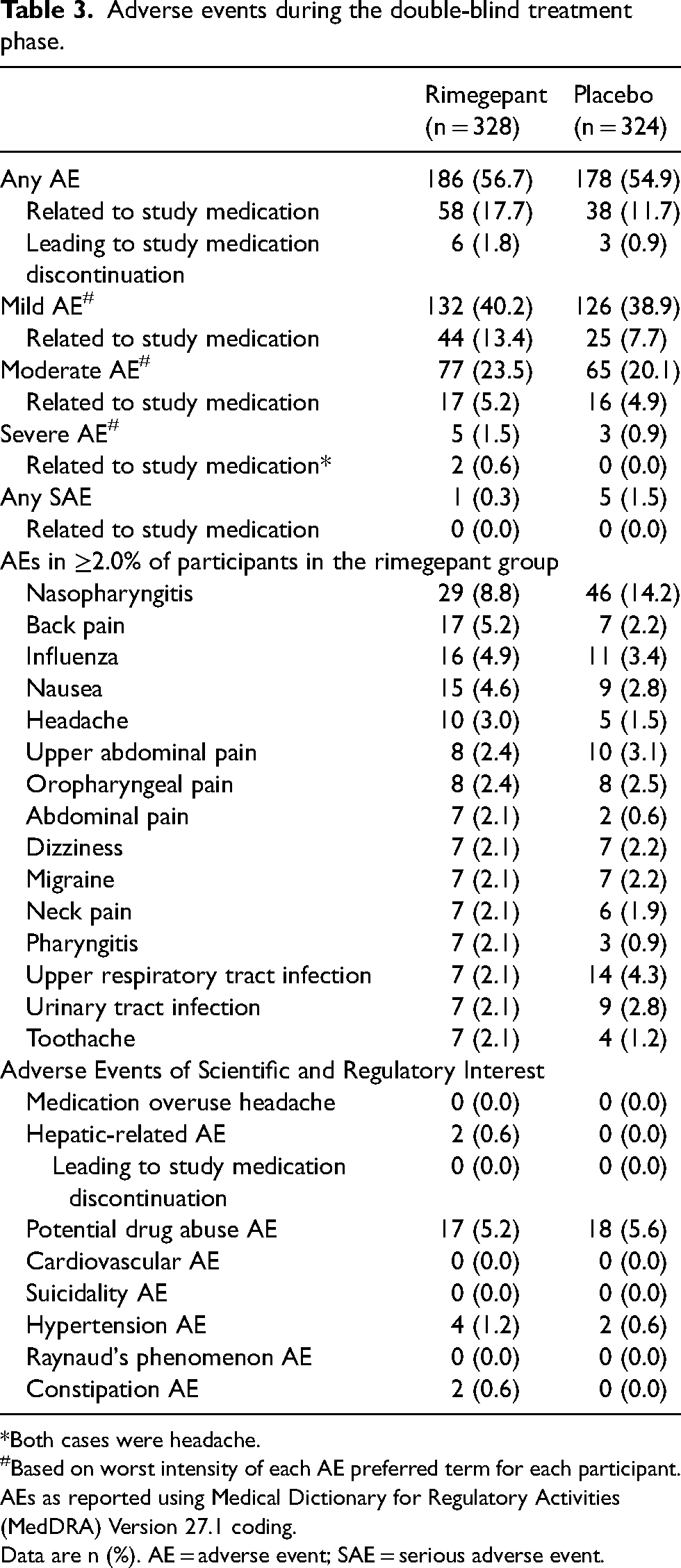
The overall incidence of AEs was similar between the rimegepant (56.7%) and placebo (54.9%) groups (Table 3), although a numerically higher proportion of participants in the rimegepant group had AEs considered by the investigator to be related to study medication (17.7% vs. 11.7%). AEs leading to study medication discontinuation were reported in 1.8% of rimegepant- and 0.9% of placebo-treated participants. The majority of AEs were considered mild or moderate in intensity. Two AEs, both of headache, were reported with severe intensity and to be related to rimegepant treatment. One participant in the rimegepant group had a serious AE of pneumonia which was not considered treatment-related.
Adverse events during the double-blind treatment phase.
*Both cases were headache.
Based on worst intensity of each AE preferred term for each participant.
AEs as reported using Medical Dictionary for Regulatory Activities (MedDRA) Version 27.1 coding.
Data are n (%). AE = adverse event; SAE = serious adverse event.
AEs reported in ≥2.0% of participants who took rimegepant were nasopharyngitis (8.8%), back pain (5.2%), influenza (4.9%), nausea (4.6%), headache (3.0%), upper abdominal pain (2.4%) and oropharyngeal pain (2.4%), as well as abdominal pain, dizziness, migraine, neck pain, pharyngitis, upper respiratory tract infection, urinary tract infection and toothache (2.1% each). The incidence of constipation was 0.6% in the rimegepant group and 0% in the placebo group. AEs evaluated by the blinded investigator as related to study medication in ≥2.0% of participants who took rimegepant were nausea (3.7%) and dizziness (2.1%).
Hypertension AEs were reported in the four (1.2%) rimegepant-treated participants; none were considered by the investigator to be related to study medication. No Raynaud's phenomenon AEs were reported during DBT phase in rimegepant-treated participants. No participants in the rimegepant group had alanine aminotransferase or aspartate aminotransferase levels elevated more than three times the upper limit of normal as well as total bilirubin more than two times the upper limit of normal. Two grade 3 laboratory findings were reported, both in the rimegepant group: one of low neutrophil count and the other of high glucose. No grade 4 laboratory findings were reported.
Discussion
This phase 4, randomized, double-blind, placebo-controlled trial is the first to rigorously demonstrate the efficacy and favorable tolerability profile of rimegepant for the preventive treatment of episodic migraine in participants with a documented history of inadequate response to multiple traditional OPMs. A particular strength of this study is the rigorous determination of prior inadequate response, ensuring that the target population was correctly selected for enrollment. This subpopulation of people living with migraine have not found effective prevention with multiple prior therapies and represent a group with a high unmet need (7,8,10,11,15,16). Previous studies have suggested this group may be less likely to experience efficacy when initiating new preventive treatments, but that CGRP-targeting therapies might offer promise (43–45).
Rimegepant is a small-molecule, oral, CGRP receptor antagonist approved in many countries as an acute treatment and preventive medication for episodic migraine (24,27). The efficacy and safety of rimegepant as a preventive therapy is supported by findings from two previous phase 2/3 clinical trials (NCT03732638 (phase 2/3) and NCT05399485 (phase 3)) (28,29). Both trials had a similar design to the current study (28-day OP and 12-week DBT phase) but were conducted at a national level (in the USA and Japan, respectively) and in slightly different populations (28,29). For example, participation criteria for the prior trials allowed enrollment of some participants with chronic migraine (i.e. 15–18 headache days per month), while the present study restricted enrollment to those with episodic migraine (less than 15 headache days per month). The prior trials also permitted concomitant use of non-CGRP-targeting preventive therapies, which were prohibited in the current study (28,29). In addition, both NCT03732638 and NCT05399485 excluded participants reporting no therapeutic response to at least two categories of preventive medication (28,29), whereas the current study specifically selected participants who had experienced inadequate response to multiple (specifically 2–4) traditional OPMs.
In the previously completed phase 2/3 trials of rimegepant for preventive treatment of migraine, the primary endpoint was change from the OP in MMDs during the last month of the DBT phase, which was a key secondary endpoint in the present study. In NCT03732638 and NCT05399485 this was achieved with a statistically significant difference −0.8 days (95% CI = −1.46 to −0.20; p = 0.0099) and −1.1 days (95% CI = −1.73 to −0.38; p = 0.0021) between rimegepant- and placebo-treated participants, respectively (28,29). These treatment differences are numerically lower than observed for the same endpoint in this study (−1.4 days; 95% CI = −2.05 to −0.80; p < 0.0001) but are in the context of numerically larger changes from the OP in both the rimegepant-treated (NCT03732638: −4.3 and NCT05399485: −2.4 vs. present study: −2.3) and placebo-treated (−3.5 and −1.4 vs. −0.9) groups (28,29). This indicates a smaller placebo response and larger therapeutic gain in the present study, which may reflect the population having more treatment experience, including previous therapies that provided inadequate efficacy. A relationship between prior treatment failures and a smaller placebo response was previously demonstrated in meta-regression of trials assessing other CGRP-targeting therapies (43,44). The primary endpoint from this study (mean change from the OP in MMDs across the entire DBT phase) was a secondary endpoint in both phase 2/3 trials with findings favoring rimegepant (p < 0.002) (28,29). In all three trials, mean change from the OP in MMDs during the first month of the DBT phase (all p < 0.001) and mean change from baseline in MSQ Restrictive Role Function at week 12 of the DBT phase (all p < 0.05) were in favor of rimegepant, and the safety profile of rimegepant was largely similar to placebo (28,29).
Several CGRP-targeting therapies have shown favorable safety and efficacy for the prevention of migraine in people with prior treatment failures (35,38,40,44,45). This includes monoclonal antibodies against the CGRP peptide, its receptor and CGRP receptor antagonists (35,38,40,44,45). These findings demonstrate the suitability of this mechanism of action for the prevention of migraine. Consensus and evidence-based guidelines from the American Headache Society, International Headache Society and European Headache Federation recommend the use of CGRP-targeting medications among first-line preventive therapies (11,14,17,18). Although several CGRP monoclonal antibodies are approved for prophylactic use, all are administered by injection, which does not align with patient preference for oral medication (19–23). Monoclonal CGRP antibodies also have long half-lives, which do not allow flexibility in treatment or ability to remove therapy rapidly if a contraindication (e.g. pregnancy or breastfeeding) or serious AE occurs (46,47).
The CGRP receptor antagonists rimegepant and atogepant are orally dosed medications approved for preventive therapy (24–27). Atogepant is approved as a once-daily tablet whereas rimegepant is approved as an ODT to be taken EOD for the prevention of episodic migraine (24–27). A recent preference study found people with migraine strongly prefer oral administration of preventive medications over injection or infusion, and felt similarly about EOD and once-daily schedules (20). Rimegepant is the only CGRP receptor antagonist approved for both acute treatment and prevention of episodic migraine and, alongside the dispersible nature of the tablet, potentially offers additional treatment flexibility (24,27).
Combined findings from the 3 trials of rimegepant EOD for the prevention of migraine demonstrate an early onset of efficacy (within the first month) that remains consistent over long-term treatment (28–30,48). This is observed in patients with and without histories of inadequate response to other preventive therapies (28–30,48). In a post hoc analysis of data from NCT03732638, it was estimated that half of participants who received rimegepant experienced a 50% reduction in weekly migraine days by week 2 of treatment, and this reduction was stable in half of all participants by week 3 (48). Therefore, rimegepant may offer a viable option to address the treatment need created by the shortfalls of traditional OPMs, which can require several months of dose titration before potential benefit is optimized and are also associated with frequent intolerance, lack of efficacy or risks of drug interaction (7,8,10–14,47,49).
The lmitations of this study include the relatively short duration of the DBT phase, which does not allow for the long-term assessment of efficacy and tolerability. Previous long-term trials with rimegepant EOD for migraine prevention suggest efficacy is enduring with a favorable safety profile (30). The ongoing OLE phase of the current study will provide longer-term treatment data from people with documented prior inadequate responses to OPMs. Because migraine trials have a significant and variable placebo response, a strength of the study is that a placebo arm was included; however, it is a limitation that an additional treatment arm was not included to directly compare against another OPM.
Conclusions
Rimegepant 75 mg ODT taken EOD was shown to be superior to placebo for the preventive treatment of episodic migraine in participants with documented inadequate response to 2–4 prior traditional OPMs. The tolerability of rimegepant was not notably different from placebo and was comparable to that observed in prior clinical trials.
Article highlights
This trial rigorously demonstrated the efficacy of rimegepant for the preventive treatment of episodic migraine in participants with a documented history of inadequate response to prior oral preventive medications.
The primary endpoint was met and statistical superiority over placebo was observed in all five key secondary endpoints.
Rimegepant demonstrated a favorable tolerability profile that was consistent with prior clinical studies.
Supplemental Material
sj-docx-1-cep-10.1177_03331024251391378 - Supplemental material for A phase 4, randomized, double-blind, placebo-controlled trial evaluating the efficacy and tolerability of rimegepant for the prevention of episodic migraine in adults with a history of inadequate response to traditional oral preventive medications
Supplemental material, sj-docx-1-cep-10.1177_03331024251391378 for A phase 4, randomized, double-blind, placebo-controlled trial evaluating the efficacy and tolerability of rimegepant for the prevention of episodic migraine in adults with a history of inadequate response to traditional oral preventive medications by Patricia Pozo-Rosich, José Antonio Gien López, Pawel Lisewski, Ayşe Neslihan Aslan, Harpreet Seehra, Alexandra Thiry, Lucy Abraham, Luz M. Ramirez, Robert Fountaine and Terence Fullerton in Cephalalgia
Footnotes
Acknowledgments
Medical writing support was provided by Jennifer Bodkin of Engage Scientific Solutions and was funded by Pfizer.
Data availability statement
Declaration of conflicting interests
The authors declared the following potential conflicts of interest with respect to the research, authorship and/or publication of this article: Patricia Pozo-Rosich has received, in the last three years, honoraria as a consultant and speaker from AbbVie, Almirall, Dr Reddy's, Eli Lilly, Lundbeck, Medscape, Novartis, Organon, Otsuka Pharmaceuticals, Pfizer and Teva Pharmaceuticals. Her research group has received research grants from AbbVie, AGAUR, EraNet Neuron, FEDER RIS3CAT, Instituto Investigación Carlos III, MICINN, Novartis and Teva Pharmaceuticals, and has received funding for clinical trials from AbbVie, Amgen, Biohaven, Eli Lilly, Lundbeck, Novartis, Pfizer and Teva. She is an associate editor for the journals Cephalalgia and Neurologia. She is the founder of ![]() , a platform to give information and tools to physicians and people who suffer from migraine and other headaches. José Antonio Gien López has received honoraria as a consultant and speaker from Grunenthal, Torrent and Pfizer. He reports no stocks or funding from any pharmaceutical company. Pawel Lisewski has nothing to disclose. Alexandra Thiry and Luz M. Ramirez were employees of Biohaven Pharmaceuticals. They are now employees of Pfizer and own stock/options. Ayşe Neslihan Aslan, Harpreet Seehra, Lucy Abraham, Terence Fullerton and Robert Fountaine are employees of Pfizer and own stock/options.
, a platform to give information and tools to physicians and people who suffer from migraine and other headaches. José Antonio Gien López has received honoraria as a consultant and speaker from Grunenthal, Torrent and Pfizer. He reports no stocks or funding from any pharmaceutical company. Pawel Lisewski has nothing to disclose. Alexandra Thiry and Luz M. Ramirez were employees of Biohaven Pharmaceuticals. They are now employees of Pfizer and own stock/options. Ayşe Neslihan Aslan, Harpreet Seehra, Lucy Abraham, Terence Fullerton and Robert Fountaine are employees of Pfizer and own stock/options.
Ethical statement
The protocol and consent forms were approved by the Institutional Review Board/Independent Ethics Committee at each site prior to initiation of the study.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship and/or publication of this article: This study was sponsored by Biohaven Pharmaceuticals, which was acquired by Pfizer, Inc., in October 2022.
Supplemental material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.