
Abstract
Aim
The aim of this trial was to evaluate the efficacy and tolerability of ubrogepant (MK-1602), a calcitonin gene-related peptide receptor antagonist (CGRP-RA), for the acute treatment of migraine.
Methods
This double-blind, placebo-controlled study randomized 834 participants to treat one migraine attack with ubrogepant 1 mg, 10 mg, 25 mg, 50 mg, 100 mg, or placebo in a 1:1 ratio. The co-primary endpoints were pain freedom and headache response at two hours. The first primary hypothesis tested the dose-response trend for two-hour pain freedom using a logistic regression model. Subsequent hypotheses tested the effects of each dose on the co-primary endpoints, using a closed sequential testing procedure to control for multiplicity.
Results
A total of 527 participants received ubrogepant and 113 received placebo. A positive response trend in the proportion of participants achieving two-hour pain freedom was demonstrated (p < 0.001). Ubrogepant 100 mg was significantly superior to placebo for two-hour pain freedom (25.5% vs 8.9%) but not for two-hour headache response. Per the prespecified multiplicity strategy, this nonsignificant result precluded further formal hypothesis testing, although the 50 mg and 25 mg doses demonstrated nominal significance over placebo for two-hour pain freedom (unadjusted p < 0.05). Overall, adverse events were similar between ubrogepant and placebo.
Conclusion
This trial supports ubrogepant’s efficacy and provides further evidence that CGRP-RAs are viable options for the acute treatment of migraine.
Introduction
Migraine represents a significant burden to patients and society (1–4). The triptans are considered the standard for acute migraine treatment and are widely used, but a substantial number of migraine sufferers do not reliably respond to them (5,6). This lack of reliable migraine control can lead to cycling through different treatment options, and is associated with both increased disability and migraine disease progression (conversion of episodic migraine to chronic migraine) (7,8). In addition, triptans, as 5-HT1B/1D receptor agonists, are potential arterial constrictors and are therefore contraindicated or recommended to be used with caution in migraine patients with comorbid cardiovascular (CV) events, conditions, and procedures (9). This cautionary group increases with age from 11% of people with migraine under 40 years, to 19% of those aged 40–59 years, and 34% of those over 60 years (9). Effective drugs with other mechanisms of action, and without CV risks and contraindications, are therefore needed.
Calcitonin gene-related peptide (CGRP) plays an important role in the pathophysiology of migraine (10–12). Various small-molecule CGRP receptor antagonists (RAs) have been studied in clinical trials and shown positive efficacy and tolerability, including proof-of-concept with olcegepant, an intravenous (IV) CGRP-RA (13), as well as oral CGRP-RAs such as BMS-927711 (14), BI 44370 TA (15), telcagepant (MK-0974) (16–19), and MK-3207 (20). CGRP RAs may be of use for those in whom triptans are not effective or well tolerated. Furthermore, CGRP-RAs are not direct vasoconstrictors (21–23), and preclinical and clinical studies suggest a low risk of CV concerns (24). CGRP-RAs may offer a migraine-specific option for patients with CV disease who cannot take triptans (5,18). This may be especially important given that physicians and migraine sufferers rate freedom from CV adverse events (AEs) as an important tolerability attribute (25).
The development programs of two small-molecule CGRP-RAs (telcagepant and MK-3207) were suspended after finding elevated levels of serum alanine aminotransferase (ALT) in a small number of participants who took the drugs on consecutive days (16,20,26). The differing pattern of findings for the two drugs (described in Ho et al. (26)) suggests that this might be an off-target effect, but the question of whether the hepatoxicity risk is a CGRP class effect is unresolved at present.
Ubrogepant (MK-1602), a novel oral CGRP-RA chemically distinct from telcagepant and MK-3207, was developed for the acute treatment of migraine. In humans, ubrogepant is absorbed rapidly (median Tmax of 0.7 to 1.5 hours) with an apparent half-life of about three hours for the α phase and about five to seven hours for the β (terminal) phase. It is metabolized by the liver, primarily via CYP3A4, and is also a p-glycoprotein substrate. The present study examined the efficacy, safety, and tolerability of ubrogepant for a single attack of migraine with or without aura in adults.
Methods
Full details of the study methods and statistical analysis are provided in the study protocol, which is available as supplementary material. Study methods are summarized below.
Participants
Key inclusion criteria included adults aged 18–65 years with a ≥1-year history of migraine with or without aura according to International Classification of Headache Disorders, second edition (ICHD-II) criteria (27) who experienced two to eight moderate or severe migraine attacks per month in each of the two months prior to screening. Key exclusion criteria included difficulty distinguishing migraine attacks from tension-type headaches; uncontrolled hypertension (controlled hypertension was allowed); basilar-type or hemiplegic migraine headache; >15 headache days per month or had taken medication for acute headache on >10 days per month in the three months prior to screening; had an acute attack within the past two months that required inpatient or emergency department treatment; had used an opioid or barbiturate for migraine in the past two months; age >50 years at the time of migraine onset; or had recently changed dose of migraine-prophylactic medication.
Study design and treatment
This was a Phase IIb, multicenter, randomized, double-blind, placebo-controlled trial (Merck Protocol MK-1602-006; ClinicalTrials.gov NCT01613248) to investigate the efficacy and safety of ubrogepant across a range of doses. The study was conducted in accordance with principles of Good Clinical Practice, and was approved by Schulman Associates Institutional Review Board Inc. Each participant provided written informed consent.
Participants were allocated in a double-blind fashion using a computer-generated randomized allocation schedule prepared by a blinded statistician. Randomization was stratified based on the participant’s self-reported usual triptan response: oral triptan high-responder (current users of triptans who respond ≥75% of the time); oral triptan low-responder (current users of triptans who respond < 75% of the time or those who have tried triptans but no longer use them); oral triptan naïve. Numbered containers were used to implement allocation. Personnel at each study site used a central interactive voice response system to determine which container should be given to which participant. All study personnel, including investigators, site staff, participants, and sponsor staff remained blinded to treatment allocation throughout the study. Unblinding took place after data collection was complete. After being randomized, participants had up to two months to treat a qualifying migraine (defined below). However, once the targeted number of treated participants with evaluable data was reached, study enrollment was stopped, as specified in the protocol at the sponsor’s discretion. Participants who were enrolled but had not treated a migraine were asked to return their study medication and were discontinued from the study.
Study medication was provided to participants in a single bottle, with a single dose per bottle, to be taken when they experienced a qualifying migraine. Eligible participants were allocated in a 1:1 ratio to one of the following treatment groups: 1, 10, 25, 50, and 100 mg of ubrogepant, or placebo. Participants were instructed to treat a qualifying migraine defined by the following conditions: migraine headache was moderate or severe (grade 2 or 3 on pain scale); migraine headache started less than four hours earlier; the migraine headache was a new headache that had not been previously treated and was not a recurrence of a previous migraine headache (at least 48 hours had elapsed since the complete termination of their last attack (pain and all associated symptoms)); no other migraine headache or headache had occurred in the previous 48 hours; the migraine headache was not already resolving on its own; prohibited medication had not been taken (see the protocol in the supplemental materials for details).
Beginning two hours after taking study medication and after the two-hour assessment had been completed, participants could take non-study rescue medication for a nonresponding headache (i.e. headache had failed to improve to grade 1/0 (mild/no pain)), or for headache recurrence.
Endpoints
Endpoints were self-reported by participants using a paper diary. Headache severity was measured on the following pain scale: 0 = none; 1 = mild; 2 = moderate; 3 = severe. Headache was assessed at baseline and then every 30 minutes up to two hours, hourly up to four hours and at 6, 8, 24, and 48 hours. The primary efficacy endpoints were pain freedom at two hours post-dose (reduction in headache severity from grade 2 or 3 at baseline to grade 0) and headache response at two hours post-dose (reduction in headache severity from grade 2 or 3 at baseline to grade 1 or 0). The secondary efficacy endpoints were: (1) absence of phonophobia, photophobia, nausea at two hours post-dose; (2) sustained pain freedom from 2–24 hours and from 2–48 hours post-dose (defined as pain freedom at two hours post-dose, with no administration of any rescue medication and no occurrence of a mild/moderate/severe headache during the respective period after dosing with the study medication); (3) sustained pain relief from 2–24 hours and from 2–48 hours post-dose (defined as headache response at two hours post-dose, with no administration of any rescue medication and with no occurrence of a moderate/severe headache during the respective period after dosing with the study medication); (4) total migraine freedom at two hours post-dose (defined as pain freedom with no photophobia, phonophobia, nausea, or vomiting at two hours post-dose); (5) total migraine freedom from 2–24 hours and from 2–48 hours post-dose (defined as sustained pain freedom with no photophobia, phonophobia, nausea, or vomiting during the specified time period).
Several exploratory efficacy endpoints were defined in the protocol but are not the focus of the current manuscript.
Safety endpoints included standard safety assessments (medical history and physical examination; vital signs; electrocardiogram; laboratory tests including hematology, chemistry, and urinalysis; and pregnancy tests for women of childbearing potential), and collection of AEs. Adjudication procedures were in place for specified vascular AEs as well as any post-screening cases of confirmed aspartate aminotransferase (AST) and/or ALT elevations ≥3 × upper-limit-of-normal (ULN).
Statistical analysis
The Full-Analysis-Set (FAS) population served as the primary population for the analysis of efficacy data. For each two-hour endpoint (pain freedom, headache response, absence of symptoms), the minimum requirement for inclusion in the FAS population was that participants were administered study treatment, had a baseline headache severity measurement, and had at least one post-dose efficacy measurement prior to, or including, the two-hour time point. The All-Subjects-as-Treated (ASaT) population was used for safety analyses.
There were three primary efficacy hypotheses: (1) There is a positive response trend across ubrogepant doses, as measured by the proportion of participants with two-hour pain freedom; (2) at least one ubrogepant dose is superior to placebo in the treatment of acute migraine, as measured by the proportion of participants with pain freedom at two hours; and (3) at least one ubrogepant dose is superior to placebo in the treatment of acute migraine, as measured by the proportion of participants with headache response at two hours.
The statistical analysis methods and multiplicity strategy are described in more detail in the protocol. In brief, the dose-response trend test on the response proportions of two-hour pain freedom was tested first and served as the gatekeeper for subsequent pairwise tests between ubrogepant doses and placebo. The closed testing procedure was then applied to each of the two co-primary hypothesis tests at significance level α = 0.05 for the comparisons of the highest dose of ubrogepant to placebo in the following order: two-hour pain freedom and two-hour headache response. The overall treatment trend test was based on a logistic regression model with continuous covariates for treatment group and age and categorical covariates for oral triptan use stratum (high responder, low responder, or naïve; see Study Design) and baseline headache severity (moderate or severe). The comparison of ubrogepant doses vs placebo were conducted using the appropriate pairwise contrasts within the logistic regression model. No trend tests were performed for the secondary endpoints, but the analysis methods were otherwise similar to those for the primary endpoints.
The planned sample size of 810 randomized participants (135 per group) had at least 84% power to demonstrate a positive dose response of two-hour pain freedom across all ubrogepant doses using a two-sided, 5% alpha-level test across a number of scenarios evaluated in a comprehensive simulation study. Power calculations were based on the assumption of a non-evaluable rate of no more than 25% and were adjusted for the planned interim analysis for futility evaluation. One interim analysis for futility was performed and reviewed by a standing internal data monitoring committee (further details in the protocol).
All statistical analyses were performed using SAS version 9.3 (SAS Institute, Cary, NC, USA).
Results
Patient characteristics
A total of 834 participants were randomized: 138 to ubrogepant 1 mg, 139 to 10 mg, 139 to 25 mg, 139 to 50 mg, 140 to 100 mg, and 139 to placebo (Figure 1). The trial was conducted from July 2012 to December 2012 at 55 study centers in the United States.
CONSORT flow diagram.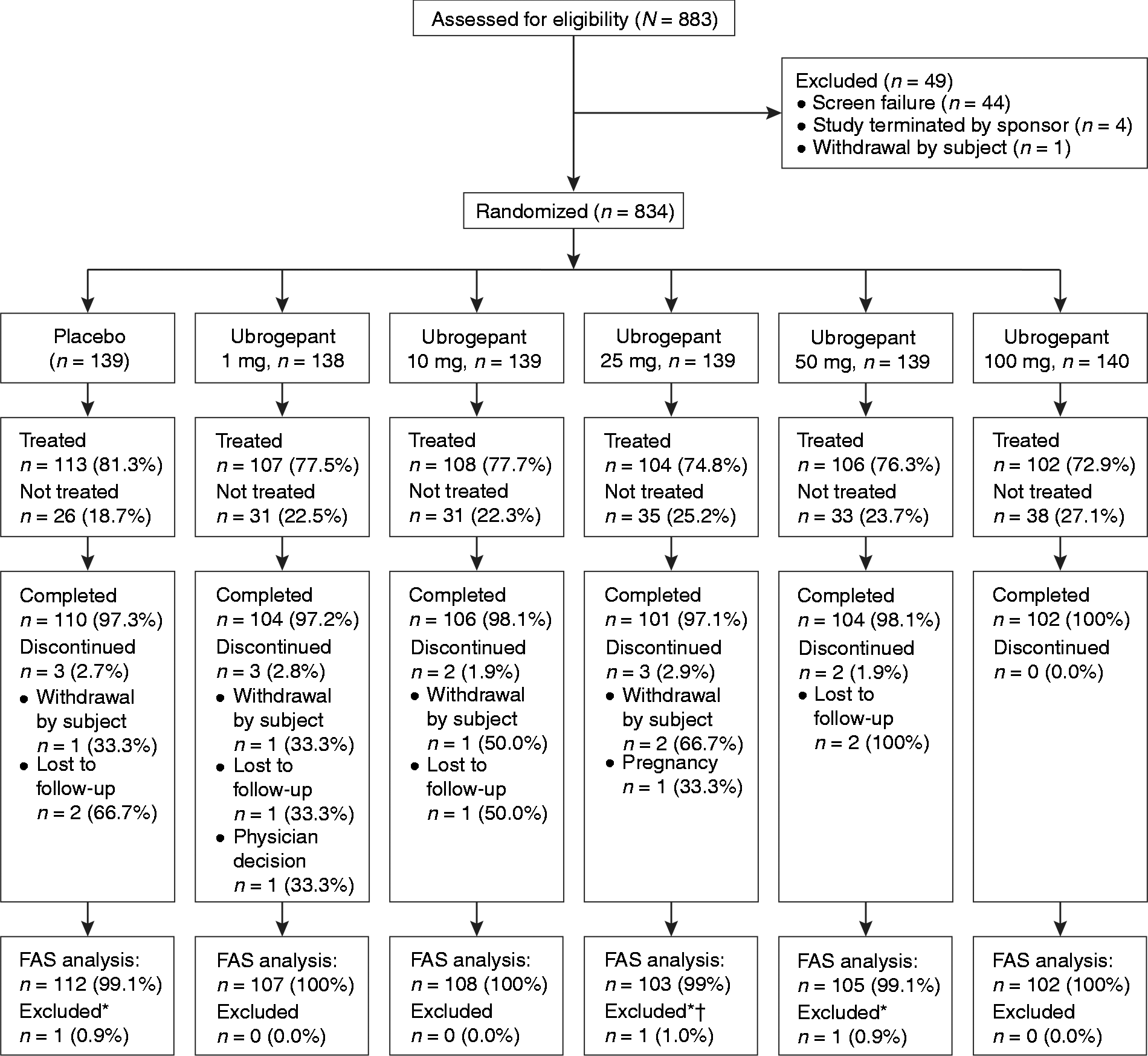
Demographics and baseline characteristics by treatment. Data are shown as mean (±SD), or absolute (n) and relative frequency (%).

BMI: body mass index; NSAID: nonsteroidal anti-inflammatory drug; SD: standard deviation.
High: current triptan user, responds ≥75% of the time; Low: current triptan user, responds <75% of the time or has tried triptans but no longer uses them; Naïve: oral triptan naïve.
Baseline characteristics of treated migraine attack.

Efficacy
Summary of primary and secondary efficacy endpoint results.

(nominal) p < 0.05, b(nominal) p < 0.01, c(nominal) p < 0.001 for ubrogepant vs placebo pairwise comparison.
CI: confidence interval.
P values computed using a logistic model adjusting for baseline severity, usual oral triptan response, and age. Numbers shown are number of treatment responders for the specific endpoint/number of participants in the endpoint-specific full analysis set population (%).
Primary endpoints
Two of the three primary hypotheses were supported. There was a positive response trend across ubrogepant doses as measured by the proportion of participants who achieved two-hour pain freedom (p < 0.001 for trend test). Pairwise comparisons were then performed for the highest dose vs placebo. Ubrogepant 100 mg demonstrated statistically significant superiority to placebo for two-hour pain freedom, supporting the second primary hypothesis. For the third primary hypothesis, two-hour headache response did not significantly differ between ubrogepant 100 mg and placebo. Per the prespecified multiplicity strategy, this nonsignificant result precluded all additional hypothesis tests from having the possibility of being formally statistically significant. However, nominal p values (i.e. unadjusted p values) were used to further characterize the results.
For two-hour pain freedom results, ubrogepant 100 mg demonstrated a statistically significantly higher success rate than placebo (25.5% vs 8.9%, p = 0.003). Ubrogepant 50 mg demonstrated a nominally significantly (i.e. results with an unadjusted p value < 0.05) higher two-hour pain-free rate than placebo (21.0% vs 8.9%, p = 0.020) as did the 25-mg dose group (21.4% vs 8.9%, p = 0.013) (Figure 2). Ubrogepant 100 mg demonstrated a higher headache response rate compared to placebo for the co-primary two-hour headache response endpoint, but this difference was not statistically significant (58.8% vs 44.6% p = 0.061) (Figure 2). None of the other ubrogepant dose groups demonstrated nominally significant superiority to placebo for the two-hour headache response endpoint.
Proportion of migraine participants reporting pain freedom at two hours post-dose, and proportion of migraine participants achieving the two-hour headache response endpoint, by treatment group.
Secondary endpoints
Ubrogepant 100 mg showed nominally significant improvements vs placebo on all secondary endpoints except absence of nausea at two hours (Table 3). Nominally significant differences from placebo were also seen on the majority of secondary endpoints for ubrogepant 50 mg (absence of photophobia at two hours, absence of phonophobia at two hours, sustained pain freedom 2–24 hours, sustained pain relief 2–24 hours, sustained pain relief 2–48 hours, total migraine freedom at two hours, total migraine freedom 2–24 hours) and on some secondary endpoints for ubrogepant 25 mg (sustained pain freedom 2–24 hours, sustained pain relief 2–48 hours, total migraine freedom at two hours). The proportions of participants meeting the pain freedom endpoint over the first eight hours after dosing (the time period over which hourly or half-hourly assessments were made) are shown in Figure 3 to illustrate the time course of ubrogepant effects. No p values are presented because this analysis was prespecified as exploratory.
Proportion of migraine participants meeting the pain freedom endpoint through eight hours post-dose, by treatment group.
Safety and tolerability
Summary of adverse events within 48 hours post-dose for the All-Subjects-as-Treated population.
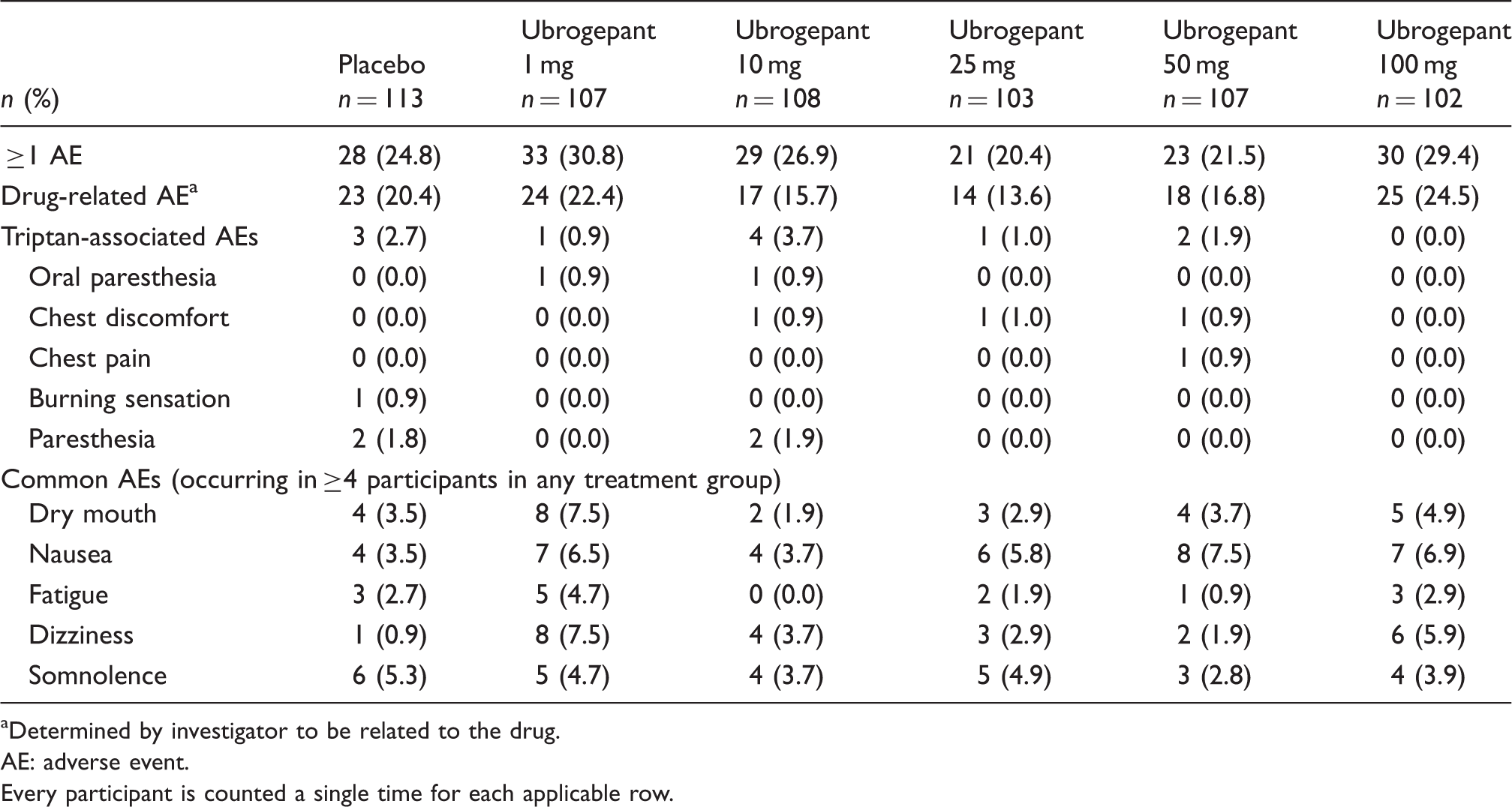
Determined by investigator to be related to the drug.
AE: adverse event.
Every participant is counted a single time for each applicable row.
Discussion
This dose-finding study assessed the efficacy, safety, and tolerability of ubrogepant for the treatment of acute migraine and found a positive response trend across ubrogepant doses, as measured by the proportion of participants who achieved the two-hour pain freedom endpoint. Participants were well balanced on baseline characteristics. The characteristics of the treated attacks did not significantly differ across the treatment groups. Ubrogepant 100 mg was statistically superior to placebo in achieving two-hour pain freedom, but did not demonstrate superiority to placebo in achieving the co-primary endpoint of headache response at two hours. Of note, the placebo response rate for headache response was relatively high (44.6%), possibly related to the 5:1 active:placebo treatment ratio, which may have created an expectancy in participants that they would receive active treatment. The sensitivity of headache response to placebo effects supports the use of the more stringent pain freedom endpoint for determining treatment effects.
In examining all efficacy data, there were consistent positive treatment effects observed for the 25-mg, 50-mg, and 100-mg doses of ubrogepant across most endpoints, suggesting that all three of these doses may be efficacious. Because formal statistical significance was achieved only for two-hour pain freedom for the 100-mg dose, the results of this study provide helpful, although not conclusive, information regarding dose selection for future clinical trials. Information from other studies could also be useful for characterizing the optimal dose(s) for future studies, such as the pharmacokinetic study of ubrogepant for the acute treatment of migraine (ClinicalTrials.gov: NCT01657370).
Given the large inter-trial variability in treatment effect sizes seen in migraine trials, it is difficult to place ubrogepant’s treatment effects in context; however, the effect sizes were generally comparable to those observed with triptans (6) and telcagepant (17,28). Ubrogepant at higher doses appeared to be particularly beneficial on sustained pain freedom measures but this single-trial observation must be replicated in future trials before drawing more definitive conclusions.
Ubrogepant was well tolerated when given acutely for a migraine attack. AE rates judged by the investigators to be related to study therapy were similar to that of placebo, with the exception of nausea and dizziness, although these events were generally mild and self-limited. The occurrence of dizziness could indicate that ubrogepant acts centrally, although it was reported only in a minority of patients (<8% in any ubrogepant group) and did not appear to be dose-related. Ubrogepant is a human p-glycoprotein substrate with moderate permeability and therefore the brain exposure is expected to be limited relative to the plasma exposure. In non-human primates, ubrogepant achieved negligible to low central CGRP receptor occupancy at plasma levels higher than those that blocked peripheral CGRP receptors in the capsaicin-induced dermal vasodilation model (unpublished data).
No significant CV events were observed and the AE profile was similar across subgroups categorized on the basis of low, moderate, or high coronary heart disease risk. However, participants with actual CV disease were not included in the study and conclusions regarding the safety of ubrogepant in such populations cannot be drawn from this single-attack study. There were no liver enzyme increases clearly attributable to ubrogepant in the present single-dose study. Further trials would be required to define the safety profile of ubrogepant for hepatic effects.
This study complied with the International Headache Society’s guidelines for controlled trials of drugs in migraine, including the use of two-hour pain freedom as an endpoint (29). The sample size was large, populations were well balanced, and demographics were reflective of the broader migraine populations, contributing to the generalizability of the trial.
Some limitations should be acknowledged. Although typical of most prior studies of drugs for acute treatment of migraine, the present study treated migraine pain and each associated symptom as independent endpoints. This approach, while rigorous, may be less sensitive to meaningful clinical benefit because profiles of associated symptoms differ from person to person and attack to attack, and because not all individuals experience all associated symptoms as equally important. A recent Food and Drug Administration draft guidance for industry on developing drugs for treatment of acute migraine (30) suggests an alternative approach. Using this approach, participants prospectively identify their most bothersome migraine-associated symptom in addition to pain. Under this approach the two co-primary endpoints would be (1) absence of headache pain at two hours post-dose, and (2) a demonstrated effect on the most bothersome migraine-associated symptom at two hours post-dose. Alternatively, a composite endpoint based on points allocated according to the presence and severity of multiple migraine symptoms (pain, nausea, photophobia, and phonophobia) could better reflect the diversity and patient-perceived importance of symptomatology experienced by migraine sufferers.
In conclusion, results from this trial provide evidence for the efficacy of ubrogepant on the two-hour pain freedom endpoint and further evidence that CGRP-RAs are viable options for the acute treatment of migraine.
Clinical implications
In this single-attack study, ubrogepant (MK-1602), a novel calcitonin gene-related peptide receptor antagonist, was found to be efficacious in the acute treatment of migraine. This study provides further evidence for calcitonin gene-related peptide receptor antagonists as being viable options for the acute treatment of migraine.
Footnotes
Acknowledgments
Medical writing assistance was provided by Steven Tresker of Cactus Communications. This assistance was funded by Merck & Co. Inc, Kenilworth, NJ, USA. Christopher Lines from Merck assisted with editing the manuscript.
Trial registration: ClinicalTrials.gov NCT01613248.
Primary investigators include: Eugene Andruczyk, Philadelphia, PA; James W Banks III, St. Louis, MO; Jo H Bonner, St. Louis, MO; Steven Bowman, Clearwater, FL; Harold Bays, Louisville, KY; Nathan L Bennett, Pittsburgh, PA; Gary D Berman, Minneapolis, MN; John J Champlin, Carmichael, CA; Harry Collins, Edison, NJ; MaryAnn Conrad, Portland, OR; Donna Marie DeSantis, Chandler, AZ; Hugh D Durrence, Charleston, SC; Victor A Elinoff, Endwell, NY; Charles Dale Eubank Jr, Corpus Christi, TX; Mark A Fisher, Oklahoma City, OK; Neil J Fraser, Troy, MI; Marshall Craig Freeman, Greensboro, NC; David L Fried, Warwick, RI; Eduard Gfeller, Maitland, FL; Gordon Leldon Gibson, Hot Springs, AR; Richard Giusti, Winter Haven, FL; Daniel B Groblewski, Jacksonville, FL; Linda Harper, Orlando, FL; Paul A Hartley, Uniontown, PA; Terence Isakov, Lyndhurst, OH; William P Jennings, S David Miller North Dartmouth, MA; Anthony D Puopolo, Milford, MA; Boris Kerzner, Baltimore, MD; Rachel Kientcha-Tita Houston, TX; Richard Krause, Chattanooga, TN; David B Kudrow, Santa Monica, CA; Kent T Kamradt, Chicago, IL; Robert B Lehman, Baltimore, MD; James G Lieber, Chandler, AZ; Gregg H Lucksinger, Austin, TX; Sean W Lynd, Cincinnati, OH; Lisa K Mannix, West Chester, OH; Herbert G Markley, Worcester, MA; Lisa Medwedeff, Plano, TX, Vishaal Mehra San Diego, CA; David J. Morin, Kingsport, TN; Clifton Scott Nicholson-Uhl, Marrero, LA; Michael Noss, Cincinnati, OH; George J Rederich, Redondo Beach, CA; Bunnie Richie, Phoenix, AZ; Jeffrey B. Rosen, Coral Gables, FL; Joel R Saper, Ann Arbor, MI; Randall J Severance, Chandler, AZ; Gioi N Smith-Nguyen, La Mesa, CA; Egilius LH Spierings, Watertown, MA; Diego T Torres, Ormond Beach, FL; Michael M Tuchman, Palm Beach Gardens, FL; Stephen R Waldman, Fullerton, CA; Jonathan Paul Wilson, Winston-Salem, NC.
Declaration of conflicting interests
The authors declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article:
TV is an employee of Merck and owns stock/stock options in Merck.
RBL has received research support from the National Institutes of Health (NIH) (PO1 AG03949 (program director, project and core leader), RO1AG025119 (investigator), RO1AG022374-06A2 (investigator), RO1AG034119 (investigator), RO1AG12101 (investigator), K23AG030857 (mentor)), the National Headache Foundation, and the Migraine Research Foundation. He serves on the editorial board of Neurology®; has reviewed for the National Institute on Aging (NIA) and National Institute of Neurological Disorders and Stroke; holds stock options in eNeura Therapeutics; and serves as a consultant, advisory board member, or has received honoraria from Allergan, the American Headache Society, Autonomic Technologies, Boehringer Ingelheim Pharmaceuticals, Boston Scientific, Bristol-Myers Squibb, CogniMed, CoLucid, Eli Lilly, Endo, eNeura Therapeutics, GlaxoSmithKline, Merck, Novartis, NuPathe, Pfizer, and Vedanta Research.
In the past 12 months, DWD has served on advisory boards and has consulted for Allergan, Amgen, Alder, Boston Scientific, CoLucid, Merck, ENeura, Eli Lilly & Company, Autonomic Technologies, Teva, Tonix, Novartis, Supenus, Scion Neurostim, Xalan, Dr Reddys, and Xenon. Within the past 12 months he has received royalties, funding for travel, speaking, or editorial activities from the following: Haymarket Media Group Ltd., SAGE Publishing, Allergan, Lippincott Williams & Wilkins, Oxford University Press, and Cambridge University Press. He receives publishing royalties for Wolff’s Headache, 8th ed. (Oxford University Press, 2009) and Handbook of Headache (Cambridge University Press, 2010).
ND is an employee of Merck and owns stock/stock options in Merck.
JYG is an employee of Merck and owns stock/stock options in Merck.
RB is an employee of Merck and owns stock/stock options in Merck.
CA is an employee of Merck and owns stock/stock options in Merck.
SKA has received grant/research support and honoraria from Allergan, Merck & Co. Inc, Teva, Amgen, and ENeura for consulting and speaking.
DM is an employee of Merck and owns stock/stock options in Merck.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: Financial support for the conduct of the research and the preparation of this article was provided by Merck & Co. Inc, Kenilworth, NJ, USA. The sponsor was involved in study design; in the collection, analysis, and interpretation of data; in the writing of the report; and in the decision to submit the article for publication.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.