
Abstract
Background
The trigeminal sensory neuropeptide calcitonin gene–related peptide (CGRP) is identified as an essential element in migraine pathogenesis.
Methods
In vitro and in vivo studies evaluated pharmacologic properties of the CGRP receptor antagonist atogepant. Radioligand binding using 125I-CGRP and cyclic adenosine monophosphate (cAMP) accumulation assays were conducted in human embryonic kidney 293 cells to assess affinity, functional potency and selectivity. Atogepant in vivo potency was assessed in the rat nitroglycerine model of facial allodynia and primate capsaicin-induced dermal vasodilation (CIDV) pharmacodynamic model. Cerebrospinal fluid/brain penetration and behavioral effects of chronic dosing and upon withdrawal were evaluated in rats.
Results
Atogepant exhibited high human CGRP receptor-binding affinity and potently inhibited human α-CGRP-stimulated cAMP responses. Atogepant exhibited significant affinity for the amylin1 receptor but lacked appreciable affinities for adrenomedullin, calcitonin and other known neurotransmitter receptor targets. Atogepant dose-dependently inhibited facial allodynia in the rat nitroglycerine model and produced significant CIDV inhibition in primates. Brain penetration and behavioral/physical signs during chronic dosing and abrupt withdrawal were minimal in rats.
Conclusions
Atogepant is a competitive antagonist with high affinity, potency and selectivity for the human CGRP receptor. Atogepant demonstrated a potent, concentration-dependent exposure/efficacy relationship between atogepant plasma concentrations and inhibition of CGRP-dependent effects.
Introduction
Migraine is a complex, chronic neurological disease that impacts over one billion people globally, and is the leading cause of years lived with disability in those under 50 years of age (1–4). Traditional oral preventive migraine treatments are often used off-label and are ineffective or poorly tolerated (5,6). Therefore, there is a need for additional preventive migraine treatments that can safely and effectively reduce the frequency and severity of migraine attacks and improve quality of life. The trigeminal sensory neuropeptide calcitonin gene–related peptide (CGRP), a potent vasodilator that is elevated during migraine attacks, has been identified as an essential element in the pathogenesis of migraine (7–9). Monoclonal antibodies (mAbs) targeting CGRP or its receptor are approved as preventive migraine treatments (8,10). Atogepant is an oral CGRP receptor antagonist that is approved by the US Food and Drug Administration for the preventive treatment of migraine in adults (11) (Figure 1). Atogepant has a plasma half-life of approximately 11 hours, making it well suited for use as a daily preventive migraine treatment (12,13). The safety and efficacy of atogepant were demonstrated in the pivotal phase 3 ADVANCE trial, where atogepant-treated participants had significantly greater decreases in mean monthly migraine days compared with placebo over 12 weeks of treatment (14).

Chemical structure of atogepant (11).
CGRP and calcitonin (CT), amylin (AMY) and adrenomedullin (AM) are collectively referred to as peptides of the calcitonin family (15,16). The receptors targeted by these peptides have been characterized and share subunits in their structures (15,16). The CGRP receptor is composed of a seven-transmembrane CT receptor (CTR)-like receptor (CLR) and the single transmembrane receptor activity modifying protein 1 (RAMP1) (17). AMY receptors are a complex of the CTR and RAMP1 (i.e. AMY1), RAMP2 (i.e. AMY2) or RAMP3 (i.e. AMY3) receptors. The AM receptors are a complex of the CLR and RAMP2 (AM1) or RAMP3 (AM2) (18). CGRP and AMY1 receptors are widely expressed in the trigeminovascular system and brainstem (19). Although CGRP can exhibit a binding affinity across this receptor family, the role of AMY and AM receptor signaling in the pathogenesis of migraine is not fully known (15).
The present study reports the characterization of the pharmacologic properties of atogepant as it relates to the pathophysiology of migraine, including in vitro and in vivo studies evaluating the pharmacologic characteristics of atogepant.
Methods
Receptor characterization studies
Receptor binding
To evaluate the affinity and selectivity of atogepant for CGRP and AMY1, receptors, cloned receptors were stably expressed in human embryonic kidney (HEK) 293 cells. The study was conducted similarly to a previously published CGRP receptor binding study for ubrogepant (20). Affinity for an endogenously expressed human CGRP receptor was assessed in a human neuroblastoma cell line (SK-N-MC membranes). Non-specific binding was determined using 10 µM MK-3207 (21), a structurally distinct CGRP receptor antagonist. Concentration–response curves were plotted to determine half-maximal inhibitory concentration (IC50) values and converted to Ki values using the equation Ki = IC50/1 + ([ligand]/Kd).
Potency at CGRP receptors
The potency of atogepant was evaluated by the inhibition of agonist-stimulated intracellular cyclic adenosine monophosphate (cAMP) levels in HEK293 cells stably expressing the human CGRP receptor.
Following agonist stimulation, cAMP concentration was measured with the homogenous time-resolved fluorescence (HTRF) cAMP Dynamic Assay (CisBio, Bedford, MA, USA). Dose–response curves were plotted and IC50 values were derived from a logistic fit as defined by the equation y = ((a – d)/(1 + (x/c)b) + d, where y is the response, x is the dose, a is the maximum response, d is the minimum response, c is the inflection point and b is the slope. For CGRP-induced changes in intracellular cAMP concentrations, Schild analysis was used to assess competitive antagonism by (DR – 1) vs. log[B], where DR is the ratio of α-CGRP half-maximal effective concentration (EC50) values in the presence and absence of atogepant and [B] is the antagonist concentration.
Receptor selectivity
To evaluate AM-induced increases in cAMP, HEK293 cells expressing the human AM1 or AM2 receptor were added to plates at 2000 cells per well and pre-incubated with various concentrations of atogepant. HEK cells stably expressing human CTR and RAMP1 were used for the AMY1 selectivity assay. For the AMY3 assay, HEK cells were transiently transfected with equal amounts of human CTR and RAMP3. To determine calcitonin-induced increases in cAMP cell lines expressing CTR, HEK293 cells with human CTR were used. Isobutyl-methylxanthine was added at a concentration of 300 µM for 30 minutes at 37°C. For AM1 and AM2, this was followed by stimulation with 1.0 nM human AM for 20 minutes at 37°C. Stimulation with 0.5 nM rat AMY and 0.2 nM human calcitonin was utilized for AMY3 and CTR assays, respectively. Following agonist stimulation, the cAMP concentration was measured with the HTRF cAMP Dynamic Assay (CisBio).
Functional characterization and efficacy studies
Anti-CGRP effect
The anti-CGRP effect of atogepant was evaluated using a capsaicin-induced dermal vasodilation (CIDV) model. Eight adult rhesus monkeys (either sex, 2.5–15.0 kg) were used for both studies. Both analyses were conducted using methodologies similar to those in previously published studies evaluating anti-CGRP effect of ubrogepant (20). Briefly, 8-mm O-rings were applied to the animals’ ventral forearm to induce dermal vasodilation, which was quantitated with laser imaging, then topical capsaicin was administered. Animals were administered three sequential intravenous (i.v.) doses of vehicle or atogepant followed by assessments of dermal blood flow and blood sampling to determine atogepant plasma levels.
Nitroglycerine (NTG) rat model of migraine
Female Sprague–Dawley rats with surgically inserted femoral vein catheters (FVCs) were used in this study. A total of 43 rats (n = 6–10 per group) were housed in an enriched environment under a 12:12 h light/dark photocycle in a climate-controlled room.
A 5 mg/ml stock solution of NTG was prepared with a buffer of ethanol, propylene glycol and sterile water. Atogepant was dissolved in a pre-made buffer of 20:20:60 (w/v) propylene glycol (PG), polyethylene glycol 300 (PEG-300) and a standardized concentrate with at least 50% phosphatidylcholine and PG (Phosal 50 PG). A behavioral model of migraine-associated hyperalgesia was induced by injecting 10 mg/kg (i.v.) of NTG every other day for 9 days via the FVCs in a volume of 2 ml/kg. The sham group received an equal dose of buffer. One hour prior to NTG injection, three groups of awake rats received oral atogepant (3, 10 and 30 mg/kg) via gavage in a volume of 1 ml/kg. Atogepant was administered once daily for 9 days.
Mechanical periorbital allodynia was measured using calibrated von Frey monofilaments 2 hours after NTG injection on days 0, 5 and 9 to determine withdrawal thresholds (22). A cutoff value was set to 15 g if no response was recorded.
Prism, version 9.1 (GraphPad Software Inc., San Diego, CA, USA) was used for statistical data analysis. Two-way analysis of variance with repeated measures and Dunnett’s post-hoc test was used to determine significant differences. p < 0.05 was considered statistically signiifcant. All data are presented as the mean ± SEM.
Safety and tolerability studies
Off-target profiling
Conventional radioligand binding and enzyme assays were performed according to Ricerca Biosciences test protocols; atogepant was tested at 10 μM in a manner consistent with previous studies (23,24).
Brain penetration
This study evaluated the brain penetration and systemic pharmacokinetics of atogepant after a single oral dose (5 and 20 mg/kg) in Sprague–Dawley rats (n = 18 per group). Plasma, cerebrospinal fluid (CSF) and whole brain (homogenates) samples were collected at 1, 2, 3, 5, 8 and 16 hours after dosing. Sample analysis for quantitation of atogepant in all samples was performed using liquid chromatography tandem mass spectrometry. Summary statistic calculations of atogepant concentrations were conducted using Phoenix WinNonlin (Certara, Princeton, NJ, USA).
Assessment of behavioral signs during chronic dosing and following abrupt withdrawal
In this study, behavioral and physical signs, including changes in body weight and locomotor activity, rearing, piloerection, as well as increased body tone, reaction to sound and irritability, were assessed during repeated (28-day, twice daily) oral administration of two doses of atogepant (10 and 30 mg/kg) or morphine (30 mg/kg, positive control) to male Sprague–Dawley rats. In addition, the above signs were evaluated following abrupt cessation of dosing for an additional 7 days to determine whether atogepant produced physical or behavioral symptoms upon withdrawal. The positive control used in this study (morphine) is known to alter the above behavioral signs after repeated dosing and following abrupt withdrawal (25).
Results
Receptor characterization studies
Receptor binding
Atogepant inhibited binding of [125I]hCGRP to cloned human CGRP receptors with a Ki (±SEM) of 0.015 ± 0.002 nM (Table 1). Similar affinity for atogepant was observed with rhesus CGRP receptors (Ki = 0.009 nM), but a weaker affinity for rat and dog CGRP receptors (Ki = 0.7 nM and Ki = 1.2 nM, respectively). Atogepant inhibited [125I]-amylin binding on membranes from cells expressing human recombinant AMY1 (Ki = 1.8 nM).
Binding affinities (Ki, nM) a of atogepant for human and non-human CGRP receptors.

CGRP, calcitonin gene–related peptide.
Values are the group mean ± SEM, n = 3–7.
Reference MK-3207 binding curves have been previously published (21).
Binding affinities were measured using [125I]CGRP and membranes from cells with native expression of the CGRP-R (rhesus monkey, rat, mouse, dog, rabbit).
Functional assays
Atogepant blocked human α-CGRP-stimulated cAMP responses in human and rhesus CGRP receptors. Atogepant did not show agonist activity on CGRP receptor-mediated cAMP concentration changes, and no significant antagonism of AM-induced cAMP stimulation of the AM1 receptor at concentrations up to 20 μM. Atogepant inhibited AM-induced cAMP accumulation in the AM2 cell line with an IC50 of 400 nM.
Atogepant inhibited AMY-induced cAMP accumulation with an IC50 of 2.4 nM on the AMY1 receptor, and an IC50 of 1418 nM on the AMY3 receptor. For the CTR, atogepant inhibited calcitonin-induced cAMP accumulation with an IC50 of 6274 nM (Table 2).
Functional potency of atogepant for human cloned CGRP and related receptors a

AM1, adrenomedullin receptor 1; AM2, adrenomedullin receptor 2; AMY1, amylin receptor 1; AMY3, amylin receptor 3; CGRP, calcitonin gene–related peptide; CTR, calcitonin receptor; IC50, half-maximal inhibitory concentration.
n = 2–9.
Calculated as (target receptor IC50 − CGRP receptor IC50)/CGRP receptor IC50.
Potency and selectivity
Atogepant blocked human α-CGRP–stimulated cAMP responses in human and rhesus CGRP receptors with a mean (±SEM) IC50 of 0.026 ± 0.005 nM and 0.045 ± 0.005 nM, respectively. The addition of 50% human serum reduced the apparent potency of atogepant by approximately 4.2-fold for the human CGRP receptor (IC50 = 0.111 ± 0.009 nM) (26). Schild regression showed that atogepant caused dose-dependent rightward shifts in the agonist dose-response curves (KB = 0.026 nM) and no reduction in the maximal agonist response.
Atogepant was highly selective for the CGRP receptor relative to the human cloned AM1 (IC50 > 18 µM), AM2 receptors (IC50 = 400 nM) and CTR receptors (IC50 = 6.3 µM). Atogepant, although selective for the CGRP receptor, displays affinity for the human AMY1 and AMY3 receptors. Atogepant blocked amylin-stimulated cAMP responses in AMY1 and AMY3 expressing cells with an IC50 2.4 nM and 1.4 µM, respectively (Table 2).
Atogepant did not show agonist activity on CGRP receptor-mediated cAMP concentration changes, and no significant antagonism of AM-induced cAMP stimulation of the AM1 receptor at concentrations up to 20 μM. These results are summarized in Table 2.
Efficacy studies
Rat NTG model of migraine
Administration of atogepant attenuated NTG-induced allodynia in a dose-dependent manner. NTG (10 mg/kg, i.v.) induced a robust periorbital mechanical allodynia 2 hours after injection (F4,38 = 45.55, p < 0.0001). This allodynia was significantly reduced by 3, 10 and 30 mg/kg of atogepant administered 1 hour prior to NTG (day 0: 30.4, 63.6 and 79.9%; day 5: 29.8, 60.0 and 85.5%; day 9: 22.6, 81.1 and 97.9%, respectively, p < 0.01) (Figure 2).

Atogepant (3, 10 and 30 mg/kg) administered to rats (n = 6–10 per group) for 9 days significantly attenuated mechanical periorbital allodynia induced by intravenous nitroglycerine 10 mg/kg. Assessments were conducted two hours after nitroglycerine injections on days 0, 5 and 9. The percent improvement in mean (SEM) von Frey withdrawal threshold for atogepant vs. vehicle is presented. *p < 0.01 vs. sham; †p < 0.05 vs. vehicle; ‡p < 0.01 vs. vehicle. F, force; g, gram; NTG, nitroglycerine; Veh, vehicle; VF, von Frey.
Efficacy in primate pharmacodynamic model
The exposure/efficacy relationship of atogepant was evaluated in a CIDV rhesus model to demonstrate the relationship between atogepant concentrations and blood flow. Doses of atogepant were chosen to provide adequate coverage of the dynamic range of the exposure/efficacy curve. A series of dose-escalating infusion protocols were employed for atogepant to determine a full dose–response relationship. Application of vehicle alone had no significant inhibition on blood flow. Atogepant was associated with a concentration-dependent inhibition in CIDV (Figure 3). However, higher plasma concentrations (>20 nM) did not appear to be associated with a greater reduction in blood flow. The exposure/efficacy relationship for inhibition of CIDV by atogepant was estimated based on data from two rhesus CIDV studies (i.v. dose range: 0.175–67 µg/kg) using a population maximum effect (Emax) model. The results suggest that atogepant has a mean EC50 of 1.04 nM (% relative standard error ∼110%), which correlates with a 90% maximal effective concentration (EC90) of approximately 9.4 nM. Overall, a predictable exposure/efficacy relationship was observed between plasma concentrations of atogepant and inhibition of dermal vasodilation.
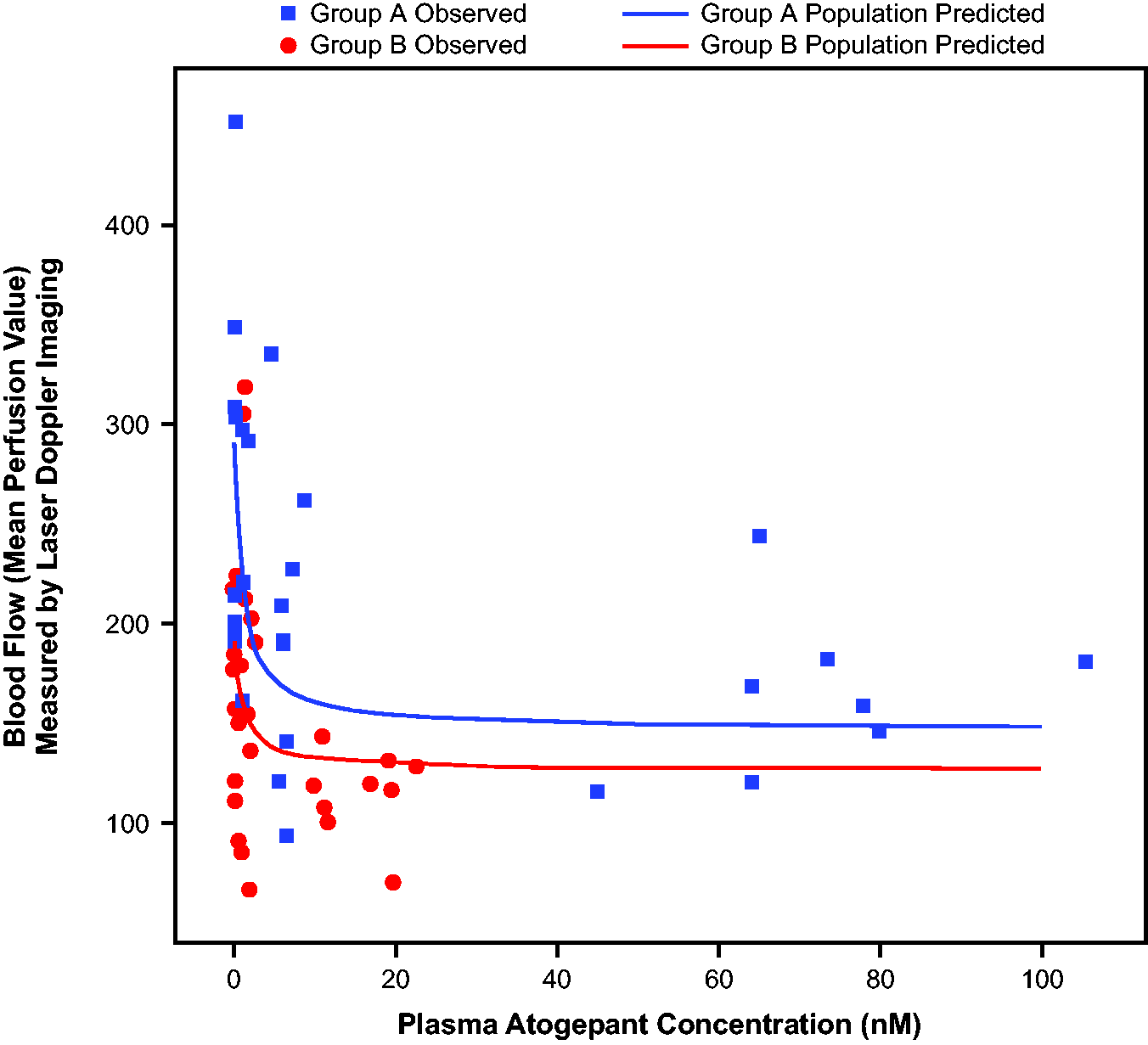
Atogepant dose-dependent inhibition of capsaicin-induced dermal vasodilation in the rhesus forearm: population model predicted vs. observed blood flow following 2 mg of capsaicin application at different plasma concentrations of atogepant. Data from eight rhesus monkeys were used in this analysis. Targeted plasma atogepant concentrations were 1, 10 and 100 nM for Group A and 0.3, 2 and 20 nM for Group B. Solid lines represent model-predicted population mean values. Each data point = single dose/measurement.
Safety and tolerability studies
Off-target profiling
Evaluation of atogepant in the off-target screen showed <50% inhibition for over 100 targets, including many known neurotransmitter receptors, enzymes or transporters, at 10 μM. Atogepant was inactive against more than 100 targets tested, including hERG (i.e. human ether-a-go-go–related gene) at 10 µM.
Brain penetration
Sprague–Dawley rats (n = 18 per group) were administered atogepant at 5 and 20 mg/kg. For both doses, only a fraction of atogepant levels measured in plasma were detected in the CSF (area under the curve from 0 to last timepoint of 16 hours [AUC0-last]: atogepant 5 mg/kg; 0.0038, atogepant 20 mg/kg; 0.0031) and brain tissue (AUC0-last: atogepant 5 mg/kg; 0.0161, atogepant 20 mg/kg; 0.0157), suggesting atogepant had minimal brain penetration (Table 3). The maximum concentration (Cmax) of atogepant in CSF and brain tissue was higher for 20 mg/kg (3.7 ± 0.2 ng/ml and 17.3 ± 5.7 ng/g) than 5 mg/kg (0.9 ± 0.3 ng/ml and 2.4 ± 0.7 ng/g). A similar dose effect was observed for the AUC0-last in CSF (5 mg/kg: 4.2 ± 1.0 ng·h/ml; 20 mg/kg: 20.0 (insufficient data available to calculate SD values) ng·h/ml) and the brain (5 mg/kg: 17.9 ± 1.4 ng·h/g; 20 mg/kg: 102.3 ± 16.3 ng·h/g). Both doses demonstrated limited brain penetration, and Cmax ratios for CSF/plasma were similar in both groups (5 mg/kg: 0.0050; 20 mg/kg: 0.0043). These data are consistent with other findings demonstrating that atogepant is a P-glycoprotein substrate and has exhibited low levels of central CGRP receptor occupancy in a primate study (data not shown).
Atogepant concentrations and concentration ratios in plasma, cerebrospinal fluid and brain tissue (Sprague–Dawley rats).

ID, insufficient data; AUC0-last, area under the curve from 0 to last timepoint of 16 hours; Cmax, maximum concentration; CSF, cerebrospinal fluid.
Reported as the mean ± SD.
Assessment of behavioral signs during chronic dosing and following abrupt withdrawal
Atogepant (10 and 30 mg/kg orally twice daily for 28 days) produced mild and transient behavioral and physical signs during the treatment phase. The plasma Cmax and AUC at the highest dose (30 mg/kg) on day 28 in this study were approximately 4.7 times and 8.6 times, respectively, the highest human dose of 60 mg. Repeated administration of atogepant (30 mg/kg twice daily) produced sporadic decreases in body weight and food intake during the course of 28-day treatment. The lower dose of atogepant (10 mg/kg orally twice daily) produced lethargy and decreased locomotor activity that were limited to week 1 only. Upon cessation of atogepant administration, no new behaviors or physical signs were reported during the withdrawal phase. By contrast, morphine (30 mg/kg, orally twice daily) produced clear behavioral and physical signs during the on-dose phase. Significant decreases in body weight and food intake were observed during week 1 but tolerance developed to these effects during week 2 onwards. Cessation of morphine administration produced further decreases in body weight and water intake during the withdrawal period.
Discussion
The analyses from these studies demonstrated that atogepant has a very high affinity and potency for the human CGRP receptor, and inhibited binding to native CGRP receptors in human neuroblastoma cell lines (SK-N-MC membranes). Radioligand binding data in different species showed that atogepant has a higher affinity for the rhesus CGRP receptor (similar to human receptors) but lower affinity for rat, mouse, rabbit and dog CGRP receptors.
A predictable exposure/efficacy relationship was observed between plasma concentrations of atogepant and inhibition of dermal vasodilation in the CIDV study. Capsaicin activates transient receptor potential vanilloid 1, producing neurogenic inflammation and vasodilation via activation of dorsal root reflexes and the release of vasoactive mediators, including CGRP (27,28). The resulting dermal vasodilation in the rhesus forearm can be blocked by CGRP receptor antagonists, thus permitting the in vivo assessment of atogepant potency against endogenously released CGRP. This finding suggests that CGRP is an important contributor to CIDV; however, other neuronal and non-neuronal transmitters such as substance P and histamine may also play a role in vasodilation, which is consistent with results from other CIDV studies for CGRP compounds (29). In the rat NTG model of facial allodynia, atogepant (3–30 mg/kg) demonstrated a dose-dependent reduction in NTG-induced facial allodynia when administered one hour prior to NTG administration.
Selectivity results among the receptor subtypes showed that atogepant is highly selective for the CGRP receptor vs. AM1 and AM2 receptors. Atogepant was also more selective for the CGRP receptor than the calcitonin, AMY1 and AMY3 receptors. Atogepant had very low levels in the brain/CSF following systemic delivery, suggesting that its mode of action is primarily outside of the CNS. Furthermore, atogepant demonstrated no physical or behavioral symptoms associated with chronic dosing and abrupt withdrawal.
Migraine is understood to be a complex, chronic disease involving dysfunction of the central and peripheral nervous systems (30). CGRP and its receptors are expressed widely throughout the nervous system (3,30). Blocking the CGRP receptors in the peripheral pathway has been demonstrated to be an effective mechanism for migraine treatment (31). The previous findings indicating that AMY and AM receptors are also activated by CGRP support the need for additional studies investigating their role in the pathophysiology of migraine (15,16). CGRP, AMY and AM receptors are known to be located throughout the nervous system, specifically on trigeminal neurons that are implicated in pain associated with a migraine attack (15,32).
These studies evaluated the affinity of atogepant for the CGRP receptor compared with other receptors in the same class. The AMY1 receptor, consisting of RAMP1 and CTR, is a second type of CGRP receptor, and has been shown to be activated by both AMY and CGRP (7). Activation of the CGRP receptor induces the signal transduction pathway involving adenylate cyclase, which raises cAMP levels, and in turn activates protein kinase A, resulting in a downstream phosphorylation cascade (7). Protein kinase A has been implicated in numerous biologic effects of CGRP, such as vasodilation and neural function (7). Similar to CGRP receptors, AMY1 is found in the trigeminal ganglion; however, its role in migraine is not yet fully understood (7,33). In one study, the AMY1 receptor was found at sites important for craniofacial pain (33), whereas, in another study, AMY receptors were found to be widely distributed throughout the nervous system and chronic AMY administration also worsened allodynia, which, taken together, suggested that the peptide has a role in pain modulation (34). A recent study found that infusion of the amylin analogue pramlintide was shown to induce migraine-like headache among patients with migraine (35). The results of the present study confirmed that atogepant is more potent at CGRP receptors (relative to AMY1); however, the plasma concentrations of atogepant attained in migraine patients likely provides inhibition of both CGRP and AMY1 signaling. For example, human atogepant plasma Cmax after the dose of 60 mg is 1192 nM. The plasma protein binding of atogepant in humans is 98.3% (26). Therefore, the unbound drug level of atogepant in the plasma at a dose of 60 mg (∼23 nM) is far above its IC50 value at the canonical CGRP receptors and about 10 times greater than its IC50 value at non-canonical AMY1 receptors. Therefore, it is expected that atogepant will produce robust inhibition of both CGRP and AMY1 signaling and negligible inhibition of AM2-mediated signaling. By contrast, mAbs targeting the CGRP (ligand or the receptor) show no measurable activity at AMY1 or other receptors in the family (36). Dual inhibition of CGRP and AMY1 receptors by atogepant may provide some additional benefit with respect to efficacy because CGRP is known to signal through both CGRP and AMY1 receptors. However, additional research is needed regarding the respective contribution of receptor subtype to more fully understand the impact of dual antagonism. The substantial cross-reactivity between AMY1 and other receptors, including those on the canonical CGRP receptor, should be further explored to determine the relevance of CGRP in the development of migraine treatments (37).
First-generation small-molecule CGRP receptor antagonists showed efficacy in clinical studies for the treatment of migraine attacks. However, some trial participants experienced liver enzyme elevations, leading to concerns regarding treatment-related hepatotoxicity and, ultimately, to discontinuation of development for these first-generation gepants (38,39). Atogepant is chemically distinct from earlier gepants and was designed with structural modifications that limit the generation of reactive metabolites (12). In several clinical trials evaluating their safety, both atogepant and ubrogepant were well tolerated with no hepatic safety issues identified (14,40,41). mAbs targeting CGRP or the CGRP receptor have been approved for the preventive treatment of migraine (12). However, mAbs require parenteral administration (subcutaneous injection or i.v. infusion) and have longer elimination half-lives compared with gepants (12). For some people with migraine, the long half-life of a mAb may not be ideal; for example, in women who are pregnant or considering pregnancy. Daily oral dosing with a gepant would allow for immediate cessation of treatment in reaction to onset of any side effects or changes in life choices.
In conclusion, the results of these studies characterized the pharmacologic profile of atogepant and demonstrated atogepant to be a potent and selective human CGRP receptor antagonist.
Clinical implications
Atogepant was found to be a competitive antagonist with high affinity, potency and selectivity for the human CGRP receptor. A clinically translatable in vivo pharmacodynamic model in rhesus monkeys demonstrated a potent, concentration-dependent exposure/efficacy relationship between atogepant plasma concentrations and inhibition of CGRP-dependent effects. Atogepant demonstrated minimal brain penetration and no adverse behavioral signs following chronic dosing or abrupt withdrawal.
Footnotes
Acknowledgments
We acknowledge Scott Pollack and Steven Gallicchio for their support and Robert Jewell (AbbVie, Irvine, CA, USA) and David Heal (RenaSci/DevelRx, Nottingham, UK) for conducting brain penetration and the chronic dosing and withdrawal studies. Allergan (prior to its acquisition by AbbVie) funded this study and contributed to the collection, analysis and interpretation of data, as well as the review and approval of the final version for publication. Medical writing support was provided to the authors by Cory R. Hussar, PhD, of Peloton Advantage, LLC, an OPEN Health company, and was funded by AbbVie. All authors had access to relevant data and participated in the drafting, review and approval of this publication. No honoraria or payments were made for authorship.
Author contributions
EM, IMB, MEF, CSB, RBW, C-CL, CPR, CS, SPMcG, GNK and PB were responsible for the study concept and design. EM, AD, SPMcG and GNK were responsible for the acquisition of data. EM, RBW, C-CL, AD, SPMcG, GNK and PB were responsible for the analysis and interpretation of data. EM, IMB, MEF, CSB, RBW, C-CL, CPR, PB, CS, SPMcG and GNK were responsible for drafting the manuscript. EM, IMB, MEF, CSB, RBW, C-CL, CPR, PB, CS, SPMcG and GNK were responsible for revising the manuscript for intellectual content. EM, IMB, MEF, CSB, RBW, C-CL, CPR, PB, CS, SPMcG and GNK were responsible for final approval of the completed manuscript submitted for publication
Data availability
AbbVie is committed to responsible data sharing regarding sponsored clinical trials. This includes access to anonymized, individual and trial-level data (analysis data sets), as well as other information (e.g. protocols, clinical study reports, or analysis plans), as long as the trials are not part of an ongoing or planned regulatory submission. This includes requests for clinical trial data for unlicensed products and indications.
These clinical trial data can be requested by any qualified researchers who engage in rigorous, independent, scientific research, and will be provided following review and approval of a research proposal, Statistical Analysis Plan (SAP) and execution of a Data Sharing Agreement (DSA). Data requests can be submitted at any time after approval in the US and Europe and after acceptance of this manuscript for publication. The data will be accessible for 12 months, with possible extensions considered. For more information on the process or to submit a request, see: ![]() .
.
Declaration of conflicting interests
Ian M. Bell, Mark E. Fraley, Christopher S. Burgey, Rebecca B. White, Christopher P. Regan and Andrew Danziger are employees of Merck Sharp & Dohme LLC, a subsidiary of Merck & Co., Inc., Rahway, NJ, USA, and own stock in Merck & Co., Inc., Rahway, NJ, USA. Eric Moore, Chi-Chung Li and Christopher Salvatore were employees of Merck Sharp & Dohme LLC, a subsidiary of Merck & Co., Inc., Rahway, NJ, USA, and own stock in Merck & Co., Inc., Rahway, NJ, USA. Chi-Chung Li is currently an employee of Genentech and may own stock. Steve P. McGaraughty, Ghazal Naseri Kouzehgarani and Pradeep Banerjee are employees of AbbVie and may hold AbbVie stock.
Funding
This study was sponsored and funded by Allergan (prior to its acquisition by AbbVie). Medical writing support was provided to the authors by Cory R. Hussar, PhD, of Peloton Advantage, LLC, an OPEN Health company, and was funded by AbbVie.