
Abstract
Background
The ongoing Pan-European Real Life (PEARL) phase 4 study is evaluating fremanezumab effectiveness and safety for the prevention of episodic and chronic migraine. This interim analysis reports primary, secondary and exploratory endpoints from when 500 participants completed at least six months of treatment.
Methods
Adults with episodic migraine or chronic migraine maintaining daily headache diaries were enrolled upon initiation of fremanezumab. Primary endpoint: proportion of participants with ≥50% reduction in monthly migraine days during the six-month period after fremanezumab initiation. Secondary endpoints: mean change from baseline across months 1–12 in monthly migraine days, acute migraine medication use, and headache-related disability. Exploratory endpoint: mean change in headache severity from baseline across months 1–12. Safety was assessed through adverse events reported.
Results
Overall, 897 participants were enrolled and 574 included in the effectiveness analyses (episodic migraine, 25.8%; chronic migraine, 74.2%). Of participants with data available, 175/313 (55.9%) achieved ≥50% monthly migraine days reduction during the six-month period post-initiation. Across months 1–12, there were sustained reductions in mean monthly migraine days, acute medication use, disability scores, and headache severity. Few adverse events were reported.
Conclusion
PEARL interim results support the effectiveness and safety of fremanezumab for migraine prevention in a real-world population across several European countries.
Trial registration: encepp.eu: EUPAS35111
Introduction
Historically, migraine preventive treatments have been hampered by non-specificity, limiting their potential for efficacy, and often leading to poor tolerability and reduced adherence (1–3). However, advancements in migraine research have led to the development of more targeted therapies, specifically monoclonal antibodies (mAb) that target calcitonin gene-related peptide (CGRP) or its receptor (4). Fremanezumab is a humanized mAb that selectively targets the CGRP ligand and is approved in Europe for migraine prevention in adults with ≥4 monthly migraine days (MMD), at 225 mg monthly, or 675 mg quarterly doses (5). A comprehensive clinical development program, comprising a series of randomized controlled trials (RCTs) in participants with chronic migraine (CM) and episodic migraine (EM), has demonstrated superiority of fremanezumab over placebo in reducing headache frequency, severity, and duration (6–12), as well as in several participant-related outcome measures (8–11).
While RCTs provide robust data supporting treatment efficacy, they often do not fully capture the complexities of real-world medical practice. Real-world evidence (RWE), therefore, has become an increasingly important complement to RCTs in guiding clinical decisions, guidelines, and shaping reimbursement policies (13). Regulatory authorities have also recognized the importance of real-world evidence in drug development and monitoring, as well as in decision-making for regulation and assessment (14). However, the currently available RWE on the effectiveness, safety, and tolerability of fremanezumab for the prevention of migraine is limited, with most studies being retrospective or conducted across only one or two countries (15–21). Therefore, large scale, prospective, multicenter, multi-country, real-world studies of fremanezumab are needed to complement the evidence generated from RCTs.
The Pan-European Real Life (PEARL; EUPAS35111) study is an ongoing phase 4 study evaluating the real-world effectiveness, safety, and tolerability of fremanezumab in a diverse European population (4). With an expected 1140 individuals with migraine followed for an observational period of 24 months, PEARL is the largest real-world study of fremanezumab to date, with a long follow-up duration. The primary objective of the study is to evaluate the effectiveness of fremanezumab in adults with chronic migraine (CM) or episodic migraine (EM) with at least four migraine days per month, as measured by the proportion of participants reaching at least 50% reduction in MMD during the six-month period after the first dose of fremanezumab in real-world clinical practice. PEARL also includes multiple secondary and exploratory objectives, which are designed to provide clinicians with solid and transferable real-world evidence on the use of fremanezumab for the prevention of migraine. Here, we present the results of the interim analysis conducted when 500 of the enrolled participants had completed six months of treatment. This analysis allows full assessment of the primary endpoint, enabling timely dissemination of real-world fremanezumab data to clinicians. Many participants also had data available for up to 12 months at the time of this analysis, which have been included for some outcomes.
Methods
The complete protocol for the PEARL study has been previously published, including all primary, secondary, and exploratory endpoints (4). Here we report the methodology used within this interim analysis.
Study oversight
The PEARL study protocol is approved by the Independent Ethics Committee/Institutional Review Board in the 11 participating European countries (Czech Republic, Denmark, Finland, Greece, Italy, Norway, Portugal, Spain, Sweden, Switzerland, and the UK), as required by local law, and all relevant local data protection laws are followed. As PEARL is a non-interventional, prospective study, no study procedures are performed beyond the participants’ care in real-world clinical practice. Informed consent is obtained from all participants as part of the study inclusion criteria; participants agree for their clinical data to be recorded anonymously and have the right to withdraw their consent at any time during the study (4).
Study design
PEARL is an ongoing, 24-month, prospective, observational, phase 4 study being conducted in 87 sites across 11 European countries. The aim of the study is to evaluate the effectiveness, safety, and tolerability of fremanezumab treatment in adult participants with EM or CM in real-world clinical practice across Europe. Other measures of clinical treatment include the impact on concomitant preventive and acute migraine medication use; treatment adherence and persistence; quality of life; severity and duration of migraine attacks; and treatment cessation and reinitiation (4).
Participants
Eligible participants are adults (≥18 years) diagnosed with CM (≥15 headache days/month for >3 months, ≥8 of which meet the International Classification of Headache Disorders criteria for migraine) or EM (≥4 MMD), who have been prescribed fremanezumab at subcutaneous doses of 225 mg monthly or 675 mg quarterly (4,22). Participants maintain a daily headache diary as part of their routine disease management; in order to be eligible, they must have ≥21 days of headache diary data in the 28 days prior to fremanezumab treatment initiation and must be willing to continue to record headache diary information throughout the study period. The published PEARL protocol contains the study’s full inclusion and exclusion criteria details (4).
The PEARL study has enrolled a total of 1140 participants, who will be followed for a 24-month observational period. The first participant was screened and enrolled in August 2020, while the last participant is expected to complete the study in March 2024. Interim analyses are scheduled for when 300, 500, and all enrolled participants have completed six months of treatment, and when all enrolled participants have completed 12 months of treatment (4,23). The final analysis is expected to be available towards the end of 2024 (4). This article presents the analyses from the second preplanned interim analysis performed after at least 500 enrolled participants completed six months of treatment, with a data cut-off of 15 February 2022, and provides RWE on fremanezumab effectiveness, safety, and tolerability. Data from the first preplanned interim analysis were previously presented at the Congress of the European Academy of Neurology 2023 (23) and have not been published as a full peer-reviewed paper.
Study procedures
Participants with migraine maintain a daily headache diary as part of their routine disease management and are required to continue doing so upon fremanezumab treatment initiation and PEARL study enrollment. Information about headache frequency, severity, duration, and characteristics, as well as information about concomitant preventive and acute migraine treatments, will ideally be captured in the participant diary entries. Accordingly, the data recorded and analyzed throughout the study period for each participant will be compared to the baseline diary recorded prior to enrollment. The headache diary also captures participant-reported outcome measures (PROMs) and validated headache-related disability tools that allows for additional consideration of the effect of fremanezumab treatment on participants’ functionality and quality of life. Throughout the PEARL study period, participants are expected to schedule physician visits every three months (±15 days) for a total of nine visits as part of routine clinical practice and disease management and at the discretion of the treating physician (Figure 1). Participants that switch from another mAb targeting CGRP or its receptor are recommended to wait until their next scheduled dose before starting treatment with fremanezumab (4).

The progress of participants through the PEARL study. CM, chronic migraine; EM, episodic migraine. Baseline is defined as the 28-day period prior to initiating treatment with fremanezumab; eligible participants have ≥21 days of data from this 28-day period in their headache diary. Fremanezumab is initiated within three months of the first visit (V0).
Assessment of outcomes
For all outcome measures, baseline data was obtained from headache diaries in the 28-day period prior to initiating treatment with fremanezumab. The primary effectiveness endpoint of the PEARL study is the proportion of participants with at least 50% reduction from baseline in average MMD during the six-month period after fremanezumab initiation. Secondary effectiveness endpoints include the mean change from baseline to months one, three, six, nine, and 12 in: MMD, the average monthly days of acute migraine medication use; and, disability scores, as measured by the Migraine Disability Assessment (MIDAS) and 6-item Headache Impact Test (HIT-6). The MIDAS questionnaire is designed to quantify migraine-related disability over a three-month recall period, with a scoring system assigned as: 0–5: Little or no disability; 6–10: Mild disability; 11–20: Moderate disability; and >20: Severe disability (24). The HIT-6 questionnaire quantifies the burden of migraine on a participant over a one-month recall period, with scoring as: 36–49: Little or no impact; 50–55: Some impact; 56–59: Substantial impact; and ≥60: Severe impact (25). Peak headache severity of remaining attacks is an exploratory endpoint and measured on an 11-point numerical rating scale (NRS) at different time points during the study period. The safety of fremanezumab treatment is evaluated based on the documentation of adverse events (AEs) reported in clinical practice (4).
Statistical analyses
Safety data are evaluated in the safety analysis set (SAS), which includes all participants who were enrolled in the study. The full analysis set (FAS) includes all enrolled participants who have ≥10 days of recorded data on the primary endpoint; effectiveness data are analyzed in the FAS using PROMs from participant diaries and other validated headache-related disability tools specified above. All variables of the PEARL study are summarized descriptively. Continuous variables are analyzed with descriptive statistics for their actual values and changes from baseline to each visit, whilst for categorical variables, frequency, and percentage will be provided. Full details of the statistical analysis can be found within the published PEARL study protocol (4). Data for primary, secondary and exploratory objectives are presented as both mean values across timepoints and as mean changes from baseline. The mean value is the average for all participants at a given time point, whereas the mean change from baseline measures the change between two time points for each individual participant. Therefore, the number of participants for the mean change is typically lower than change from baseline as data must be available for the individual participants at both time points.
Results
Study population
At data cut-off for this preplanned interim analysis, 897 participants were enrolled. Most were from Italy (n = 304), followed by Sweden (n = 105), Switzerland (n = 100), and the Czech Republic (n = 93) (Online Supplementary Table 1). All participants were included in the SAS and 574 were included in the FAS. Participants were excluded from the FAS due to missing baseline data; discontinuation due to protocol deviation; insufficient migraine days in the observation period or the baseline period; and/or no diary data (Figure 2). The population included in the study are varied and reflective of the real-world, with many participants having comorbidities and using concomitant medications (Table 1). Participants had a mean age of 45.2 years; 88.5% were female; 93.6% were white, 0.2% were Asian, race was not reported for 6.3%; and there was a 3:1 ratio of participants with CM (74.2%) versus EM (25.8%). Common comorbid conditions included psychiatric disorders (reported in 27.2% of participants), such as depression (13.8%) and insomnia (5.2%); musculoskeletal disorders (17.4%); and gastrointestinal disorders (16.7%).

Participant inclusion process. FAS, full analysis set.
Participant demographic and baseline characteristics.

BMI, body mass index; CM, chronic migraine; EM, episodic migraine; FAS, full analysis set; SD, standard deviation.
Includes enrolled participants with ≥10 days of diary entry data post-treatment initiation.
Total participants with medical history (n = 459).
Includes system organ class reported in ≥10% of the study population.
Includes specific disorders reported in ≥2% of the study population.
Some participants can have several different comorbidities and thus, the numbers do not sum up to the total number of participants.
Other disorders include participants who had several comorbidities.
Participants had a mean disease duration of 26.2 years from initial symptom onset, and 16.6 years from official diagnosis, until fremanezumab initiation (Table 1). Anticonvulsants, beta-blockers, and tricyclics were the three most common prior migraine preventive treatment classes used by participants with both EM and CM, with anticonvulsants being the most used in EM (76.4% of participants) and beta-blockers the most used in CM (61.7%). A total of 66 participants (11.5%) had previously taken erenumab as preventive migraine therapy and two participants (0.3%) had previously used galcanezumab (these two participants had used both erenumab and galcanezumab). The duration of prior preventive migraine therapy and reasons for stopping treatment were recorded and are available in the supplementary section (Online Supplementary Figure 1).
Primary, secondary and exploratory endpoints
As expected in a real-world study, the compliance of participants in filling in the diaries and the other tools for capturing the secondary and exploratory endpoints was lower than RCTs, and data collected were based on features captured in diaries prior to enrollment. As a result, at cut-off not all data were available for each endpoint and missing data have been excluded. This is reflected in the dropping participant numbers for each timepoint.
Responder rate
A total of 313 participants had data for the primary endpoint: 72 (23.0%) with EM and 241 (77.0%) with CM. Overall, 55.9% of participants (175/313) had ≥50% reduction in MMD during the six-month period after fremanezumab initiation. A ≥50% response rate was recorded in 69.4% (50/72) of participants with EM and 51.9% (125/241) of participants with CM (Figure 3). The proportions of participants with ≥50% reduction in MMD were also recorded at months one, three, six, nine, and 12 after fremanezumab initiation; over half of participants with EM and CM had a ≥50% reduction in MMD from their baseline at all timepoints from month 3–12 (Online Supplementary Figure 2).
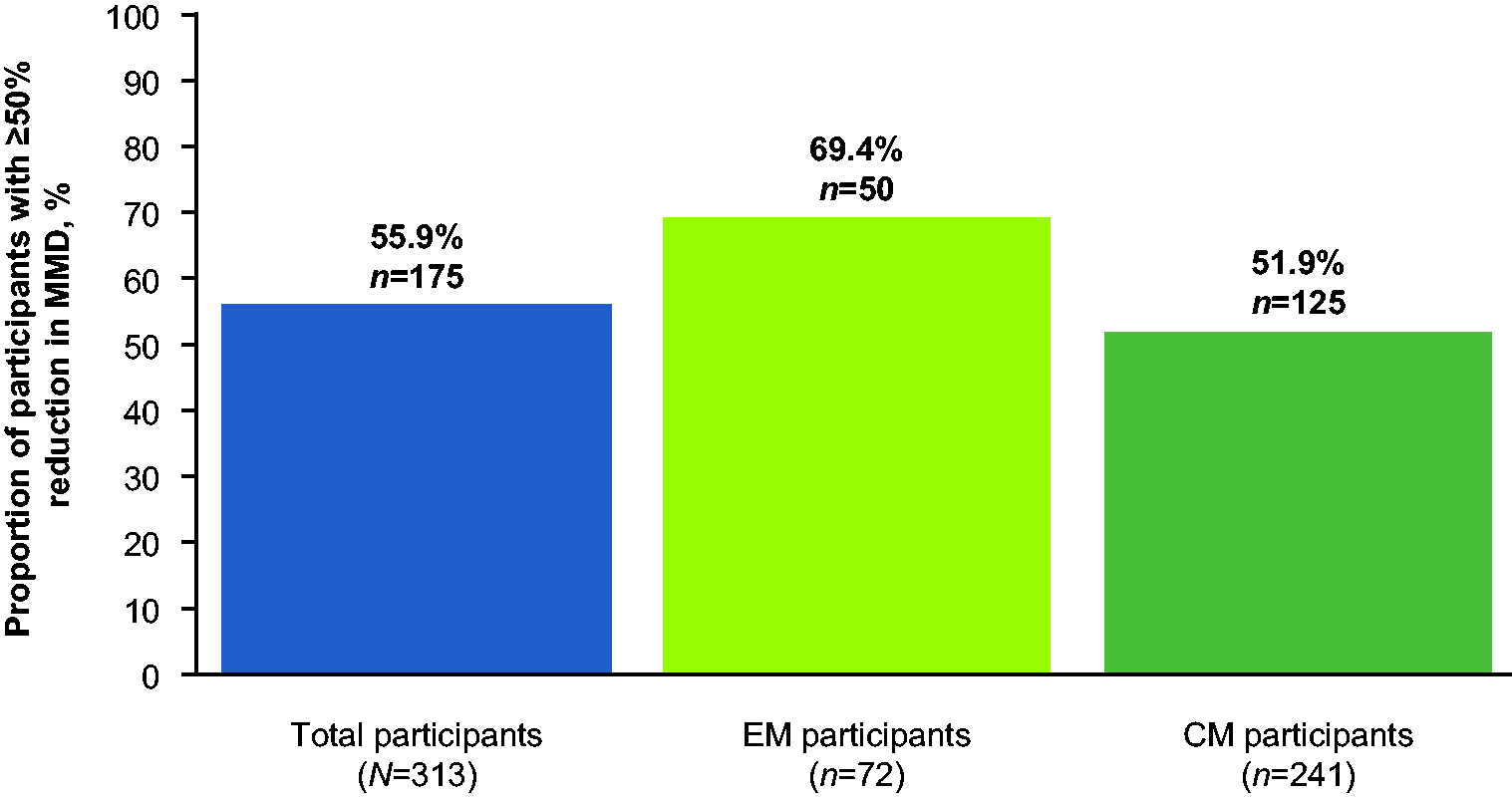
Proportion of participants with ≥50% reduction in MMD during the six months after fremanezumab initiation by migraine type (Primary endpoint). CM, chronic migraine; EM, episodic migraine; MMD, monthly migraine days. At cut-off, not all data for this endpoint were available and missing data have been excluded.
Monthly migraine days
At baseline, the mean MMD for all participants was 14.8. At months one, three, six, nine and 12 after fremanezumab initiation, this reduced to 8.9, 7.6, 6.7, 6.0, and 6.4 days, respectively. Considering MMD change from baseline, this reduced in participants with EM from a baseline of 10.1 MMD to 5.3, 4.0, 3.8, 3.7, and 2.3 at months one, three, six, nine, and 12, whilst participants with CM had a baseline of 16.5 MMD, declining to 10.1, 8.8, 7.6, 6.5, and 6.9 (Figure 4). The mean reduction from baseline in MMD at month six was –8.0 for all participants, –5.7 for EM participants, and –8.7 for CM participants. For all participants, the mean reduction from baseline was –8.5 days at month nine and –8.3 days at month 12.

Mean change from baseline in MMD at months one, three, six, nine, and 12 by migraine type (secondary endpoint). CM, chronic migraine; EM, episodic migraine; MMD, monthly migraine days; SD, standard deviation.At cut-off, not all data for this endpoint were available and missing data have been excluded.
Acute migraine medication use
At baseline, the mean number of days with acute migraine medication use was 11.1 per month for all participants. This decreased to 5.3, 4.6, 4.1, 3.9, and 4.3 days at months one, three, six, nine, and 12, respectively. In EM participants, a baseline of 9.2 days declined to 4.0, 3.1, 2.9, 3.5, and 1.1 days at months one, three, six, nine, and 12, and in CM participants a baseline of 11.9 days declined to 5.8, 5.0, 4.4, 4.0, and 4.7 days. The average change from baseline was consistent through to month 12, ranging from –5.6 to –6.7 for the total participant population (Figure 5).

Change from baseline in mean monthly days of acute migraine medication use at months one, three, six, nine, and 12 by migraine type (secondary endpoint). CM, chronic migraine; EM, episodic migraine; SD, standard deviation. At cut-off, not all data for this endpoint were available and missing data have been excluded.
Migraine-related disability
At baseline, month six, and month 12, respectively, mean MIDAS scores were 87.2, 27.9, and 27.3 for all participants; for participants with EM they were 66.3, 6.0, and 7.2; and for participants with CM they were 92.5, 34.5, and 30.8. Similarly, mean HIT-6 scores were 66.2, 56.2, and 55.4 for all participants; 66.3, 52.5, and 47.5 for EM; and 66.1, 57.2, and 56.9 for CM, respectively, at baseline, month six, and month 12.
The mean changes from baseline in MIDAS and HIT-6 scores were consistent through to month 12, with slightly higher reductions in HIT-6 score observed in participants with CM than those with EM (Figure 6).

Change in disability scores from baseline to months three, six, nine, and 12 by migraine type: (A) MIDAS score and (B) HIT-6 scorea (secondary endpoint). CM, chronic migraine; EM, episodic migraine; HIT-6, 6-item Headache Impact Test; MIDAS, Migraine Disability Assessment; SD, standard deviation. aHIT-6 possible score ranges from 36-78, and MIDAS possible score ranges from 0-270, with higher scores indicating more severe disability for both scales. At cut-off, not all data for this endpoint were available and missing data have been excluded.
Peak headache severity
In the total population, mean participant-reported peak headache severity of headache attacks at baseline was 6.1 on an 11-point numerical rating scale (NRS). Baseline mean peak headache severity was slightly higher for participants with CM (6.3/11) than EM (5.6/11). Headache severity decreased after fremanezumab initiation, with mean scores of 5.2, 5.1, 4.7, 5.0, and 4.9 at months one, three, six, nine, and 12, respectively (Online Supplementary Figure 3). At months one, three, six, nine, and 12, respectively, NRS values dropped from 6.3 to 5.3, 5.3, 5.1, 5.0, and 5.2 in CM and from 5.6 to 4.9, 4.6, 4.0, 5.0, and 3.5 in EM.
Mean change from baseline was –1 at month one and remained stable through to month 12.
Safety
Of 897 participants in the SAS, 25.0% of participants (n = 224) experienced ≥1 AE and 17.7% (n = 159) experienced ≥1 AE that had been classified by the investigator to be related to fremanezumab treatment (Table 2). The most common AEs, reported in ≥3% of participants, were injection-site erythema (5.4% [48/897]), injection-site pruritus (3.3% [30/897]), and constipation (3.1% [28/897]). Excluding one case of constipation, all reported cases of these common AEs were classified as related to fremanezumab treatment. One participant experienced a drug-related serious AE: dysphonia, and 2.2% of participants discontinued treatment due to AEs, with most citing adverse events such as drug ineffectiveness (n = 6), injection site erythema (n = 5), and injection site pruritus (n = 4).
Safety analysis.

AE, adverse event.
Drug-related AEs have been classified by the investigator to be related to fremanezumab treatment.
Includes system organ class reported in ≥5% of the study population.
Includes specific disorders reported in ≥2% of the study population.
Discussion
This interim analysis of the PEARL study was conducted following the completion of six months of fremanezumab treatment by 500 adults with migraine. The FAS incorporated 574 participants, predominantly female with the mean age of 45.2 years. Three quarters had CM and one quarter had EM. Participants had previously failed multiple migraine preventive treatments and had a substantial burden of migraine-related disability and psychiatric comorbidities. Within the first six-month period following fremanezumab initiation, participants reported a multidimensional improvement in headache symptoms, encompassing a reduction in MMD, acute migraine medication use, peak headache severity, and disability scores.
While early RWE data on the effectiveness, safety, and tolerability of fremanezumab were collected retrospectively on a relatively small number of participants (16), prospective studies have followed, enrolling larger cohorts for a longer duration. For example, the FRIEND study was conducted across 28 Italian headache centers and demonstrated the effectiveness of fremanezumab in reducing MMD and other effectiveness endpoints through week 24 in 148 participants with high-frequency EM and CM who were not previously exposed to antibodies targeting CGRP (18). In contrast, PEARL includes participants from 11 European countries with diverse demographics, generating the largest real-world data pool available for fremanezumab from participants with varied ages, nationalities, ethnic backgrounds, comorbidities, and concomitant medications. Most participants included in this analysis were receiving a concomitant medication and many had comorbid conditions, reflecting the complexity of the migraine patient population in the real-world and underscoring the relevance of this study to everyday practice. Other real-world studies are being conducted on the effectiveness of fremanezumab in the prevention of migraine; however, they are not yet available as full peer-reviewed papers, preventing a critical analysis in relation to the findings presented here (19,20).
The findings presented in this interim analysis corroborate the efficacy data from preceding RCTs where fremanezumab demonstrated a maintained superiority over placebo. Indeed, the effectiveness results of this interim analysis, in terms of the primary and secondary endpoints, are numerically higher than those observed in fremanezumab RCTs (6–11). These findings expand on evidence provided by the phase 3b FOCUS RCT, in which greater reductions in MMD over a 12-week study period were observed with quarterly or monthly fremanezumab versus placebo in participants who had documented inadequate response to two to four classes of prior migraine preventive treatment (9). The diverse population enrolled in the PEARL study further expands fremanezumab data available for participants for whom prior preventive treatments have failed, reinforcing clinician decisions.
A critical consideration in any therapeutic choice is drug tolerability, as it directly influences adherence and persistence. Due to the varied nature of real-world patient populations, these studies may identify additional AEs to those observed in pivotal RCTs. For example, a 52-week observational trial of erenumab in adults with CM reported constipation as the most common AE (affecting 41.3% of participants), as well as the most common reason for treatment discontinuation due to lack of tolerability in 7.3% of all participants (26). The AEs reported in this real-world study of erenumab were substantially higher than observed in erenumab RCTs. Additionally, post-marketing data identified an association between erenumab and elevated blood pressure, leading to hypertension being added to the Warnings and Precautions section of the prescribing information (27). Previously published real-world fremanezumab data have not revealed any new safety signals (16,18), and this interim analysis of PEARL reported few AEs with fremanezumab; one participant experienced a drug-related serious AE (dysphonia), and 2.2% of participants discontinued treatment due to AEs. Fremanezumab-related constipation was reported as an adverse event in 3.0% of participants; this is markedly lower than with other mAbs targeting CGRP or its receptor observational study results. These PEARL safety findings have no disparities with previous fremanezumab RCTs, confirming the manageable tolerability profile of fremanezumab in real-world population receiving various concomitant medications.
There are some limitations that need to be acknowledged regarding the PEARL study. First, the outcomes reported in participant headache diaries relies on the accuracy of the recorder and is subject to human error. In addition, the real-world nature of the study increases the percentage of missing data compared to an RCT, although this is an inherent challenge in RWE collection (28). As a result, data collection and comparisons taken at month six and beyond should be interpreted with caution as sample sizes may become thinner. However, despite these limitations, PEARL remains the largest prospective study of fremanezumab, with a greater number of participants than pivotal RCTs (8–10). As the study progresses, more data will become available in future interim and final analyses, which will include a larger and more representative population of participants, ensuring more robust evidence.
Conclusion
This interim analysis of PEARL confirms the efficacy and safety of fremanezumab for migraine prevention in a real-world setting in the largest migraine population to date, from diverse European countries. The present findings complement the evidence supporting the use of fremanezumab in migraine prevention generated in RCTs.
Article highlights
PEARL represents the most extensive prospective, observational, multinational study of fremanezumab to date. Following the initiation of fremanezumab treatment, participants with EM and CM reported meaningful reductions in MMD, acute migraine medication use, peak headache severity, and improvements in disability scores. Adverse events were minimal and similar to those observed in RCTs with fremanezumab, leading to treatment discontinuation in only 2.2% of participants.
Supplemental Material
sj-pdf-1-cep-10.1177_03331024231214987 - Supplemental material for Real-world effectiveness of fremanezumab for the preventive treatment of migraine: Interim analysis of the pan-European, prospective, observational, phase 4 PEARL study
Supplemental material, sj-pdf-1-cep-10.1177_03331024231214987 for Real-world effectiveness of fremanezumab for the preventive treatment of migraine: Interim analysis of the pan-European, prospective, observational, phase 4 PEARL study by Messoud Ashina, Dimos D. Mitsikostas, Faisal Mohammad Amin, Pinar Kokturk, Christoph J. Schankin, Gurdal Sahin, Patricia Pozo-Rosich, Paul J. Dorman, Tomáš Nežádal, Anne Christine Poole, Isabel Pavão Martins, Marja-Liisa Sumelahti, Verena Ramirez Campos, Andrew H. Ahn, Leonidas Lyras and Cristina Tassorelli in Cephalalgia
Footnotes
Acknowledgments
Medical writing and editorial support for the development of this manuscript, under the direction of the authors, was provided by Molly Clark, Sophie Roberts, and Olivia Morris of Ashfield MedComms, an Inizio company, and funded by Teva Pharmaceuticals.
Data availability statement
The data sets used and/or analyzed for the study described in this manuscript are available upon reasonable request. Qualified researchers may request access to patient level data and related study documents including the study protocol and the statistical analysis plan. Patient level data will be de-identified and study documents will be redacted to protect the privacy of trial participants and to protect commercially confidential information. Please visit ![]() to make your request.
to make your request.
Author contributions
MA and PK conceptualized and designed the study, analyzed the data, and reviewed the manuscript. FMA and AHA contributed to the conceptualization and design of the study and reviewed the manuscript. DDM, VRC, LL and CT analyzed the data and reviewed the manuscript. CJS, GS, PPR, PJD, TN, ACP, IPM and MLS reviewed the manuscript.
Declaration of conflicting interests
The authors declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: MA reports personal fees and PI in clinical trials for AbbVie, Amgen, Eli Lilly, Lundbeck, Novartis, Pfizer, and Teva Pharmaceuticals; and research grants from Lundbeck Foundation, Novo Nordisk Foundation, and Novartis. DDM has received honoraria, research, and travel grants from Allergan/Abbvie, Amgen, Biogen, Cefaly, Genesis Pharma, Electrocore, Eli Lilly, Lundbeck, Merk-Serono, Mertz, Novartis, Roche, Sanofi, Specifar, and Teva Pharmaceuticals; participated in clinical trials for Amgen, Cefaly, Electrocore, Eli Lilly, Genesis Pharma, Lundbeck, Mertz, Novartis, Specifar, and Teva Pharmaceuticals as principal investigator; and is President of the board of the Hellenic Headache Society and Co-chairman of the management group of the Headache Panel at the European Academy of Neurology. FMA reports lectures, advisory boards, and/or PI in clinical trials for Eli Lilly, Lundbeck, Novartis, Pfizer, and Teva Pharmaceuticals. PK, VRC, and AHA are employees and/or shareholders of Teva Pharmaceuticals. LL is a former employee of Teva Pharmaceuticals. CJS reports consulting, advisory boards, presentations, and travel support from Novartis, Eli Lilly, Teva Pharmaceuticals, AbbVie, Allergan, Almirall, Amgen, Lundbeck, MindMed, and Grünenthal; is a part-time employee of Zynnon; and reports research support from the Swiss Heart Foundation, Eye on Vision Foundation, Baasch-Medicus Foundation, and German Migraine and Headache Society. GS reports advisory boards and/or PI in clinical trials for AbbVie, Lundbeck, Novartis, Pfizer, and Teva Pharmaceuticals; and research support from Vinnova (Sweden’s innovation agency), Lund University, and the Swedish Neurological Association. PPR reports grant/research support from AbbVie, AGAUR, EraNet NEURON, Instituto Investigación Carlos III, MINECO, Novartis, RIS3CAT FEDER, and Teva Pharmaceuticals; and consultancy or education for AbbVie, Amgen, Biohaven, Chiesi, Eli Lilly, Lundbeck, Novartis, Pfizer, and Teva Pharmaceuticals. PJD reports educational meetings and/or collaboration in clinical trials for Allergan/AbbVie, electroCore, Novartis, Teva Pharmaceuticals, and Lundbeck. TN reports consulting, speaking fees, and travel grants from Eli Lilly, Glenmark, Lundbeck, Novartis, Pfizer, St. Jude Medical, Teva Pharmaceuticals, and UCB; and advisory boards or PI in clinical trials for AbbVie, Amgen, Eli Lilly, Lundbeck, Neurocrine, Novartis, Teva Pharmaceuticals, and UCB. ACP reports lectures, advisory boards, and/or PI in clinical trials for Allergan, AbbVie, Pfizer, Teva Pharmaceuticals, Novartis, Lundbeck, Eli Lilly, and Roche. IPM reports lectures, advisory boards, and/or PI in clinical trials for Allergan-AbbVie, Teva Pharmaceuticals, Novartis, Lundbeck, and Eli Lilly. MLS reports lectures, advisory boards, and/or PI in clinical trials for Pfizer, Teva Pharmaceuticals, Novartis, Lundbeck, AbbVie, and Eli Lilly. CT reports personal fees for the participation in advisory boards or symposia for AbbVie, Dompé, Eli Lilly, Lundbeck, Novartis, Pfizer, and Teva Pharmaceuticals; she reports institutional fees for conducting clinical trials for AbbVie, Dompé, Eli Lilly, Lundbeck, Novartis, Pfizer, and Teva Pharmaceuticals; she received funding for research projects from the European Commission, the Italian Ministry of Health, the Migraine Research Foundation, and the Italian Multiple Sclerosis Foundation.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This study was funded by Teva Pharmaceuticals.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.