
Abstract
Background
Published evidence supporting efficacy of calcitonin gene-related peptide receptor antagonists as acute migraine treatments in males is limited.
Methods
To fill the gap, we present male and female data from four ubrogepant and four atogepant randomized, double-blind, placebo-controlled trials for acute and preventive treatment of migraine, respectively. Acute outcomes included 2-h pain freedom and absence of most bothersome symptom (co-primary; headache-phase randomized, double-blind, placebo-controlled trials); absence of moderate-to-severe headache within 24 h (primary; prodrome randomized, double-blind, placebo-controlled trial). Preventive outcome included change from baseline in mean monthly migraine days across 12 weeks (primary).
Results
Pooled data from phase 3 headache-phase ubrogepant randomized, double-blind, placebo-controlled trials showed similar rates of pain freedom (19.4% vs 21.1%) and absence of most bothersome symptom (35.1% vs 39.0%) 2 h post-dose between males and females, respectively. Time course of pain freedom and absence of most bothersome symptom over 48 h was similar between male and female subgroups. Comparable reductions in mean monthly migraine days across 12-week treatment periods were found between males and females treated with atogepant 60 mg once-daily in pooled episodic migraine and chronic migraine randomized, double-blind, placebo-controlled trials.
Conclusion/Interpretation
In ubrogepant and atogepant randomized, double-blind, placebo-controlled trials, although analysis power for males is limited due to small sample sizes, evidence supports similar treatment effects in males and females with migraine.
Trial registration
ClinicalTrials.gov: NCT02828020; NCT02867709; NCT04492020; NCT01613248; NCT02848326; NCT03777059; NCT04740827; NCT03855137
Introduction
Migraine affects >1 billion people worldwide (1) and is the second leading cause of years lost to disability, negatively affecting individuals’ overall health, quality of life, and functioning (2,3). The prevalence of migraine is higher in females (17.0%) than males (8.6%) (1). Females with migraine often experience more frequent and longer attacks (4,5). Because of these well-recognized differences, randomized, double-blind, placebo-controlled trials (RCTs) of migraine therapeutics, including calcitonin gene–related peptide (CGRP) receptor antagonists, have enrolled more females than males (6).
Ubrogepant and atogepant are oral, small-molecule CGRP receptor antagonists approved for the acute (7–10) and preventive (11–14) treatment of migraine, respectively. Efficacy data from trials of CGRP receptor antagonists in broad adult populations that included both males and females have demonstrated benefit in the acute and preventive treatment of migraine (15,16). Based on an incomplete review of RCT data, it has been suggested that evidence supporting the therapeutic benefit of CGRP receptor antagonists for “short-term” (i.e., single dose) treatment of acute migraine attacks in males is lacking (17). A subsequent review concluded that there is a clear benefit for CGRP-targeted treatments (including gepants) for the preventive treatment of migraine in males but available clinical evidence is insufficient to support the effectiveness of CGRP receptor antagonists (ubrogepant, rimegepant, and zavegepant) in males for acute treatment (18). To address the important scientific and clinical question of whether CGRP receptor antagonists have efficacy in male patients with migraine, a comprehensive review of prespecified endpoints from four ubrogepant and four atogepant RCTs was conducted, using the entirety of data available to the authors.
Methods
Study designs
These analyses were conducted with data from four ubrogepant trials: a phase 2b single-attack, headache-phase RCT (P006 [NCT01613248] (9)); two phase 3 single-attack, headache-phase RCTs (UBR-MD-01 [NCT02828020] (7); UBR-MD-02 [NCT02867709] (8)); and a phase 3 prodrome (premonitory) phase crossover RCT (3110-304-002 [NCT04492020] (10)). The four atogepant trials included a phase 2b/3 episodic migraine RCT (CGP-MD-01 [NCT02848326] (11)), a phase 3 episodic migraine RCT (3101-301-002 [NCT03777059] (12)), a phase 3 episodic migraine RCT in individuals who were previously failed by 2–4 classes of oral preventive migraine treatment (3101-304-002 [NCT04740827] (14)), and a phase 3 chronic migraine RCT (3101-303-002 [NCT03855137] (13)). An overview of the designs of these clinical trials is provided in Online Supplemental Table S1. All primary and secondary endpoints assessed in these analyses were prespecified.
All trials were conducted in accordance with the Declaration of Helsinki. Each participating institution obtained approval from the relevant ethics committee or institutional review board (7–14). All participants provided written informed consent prior to the initiation of any study procedures.
Ubrogepant
The ubrogepant single-attack, headache-phase RCTs enrolled adults with ≥1-year history of migraine, including 2–8 moderate-to-severe migraine attacks per month. Randomized participants had up to 60 days to treat a migraine attack with moderate-to-severe headache or were withdrawn. The sex subgroup analyses in this manuscript focused on the approved doses of ubrogepant 50 mg and 100 mg. The prodromal phase study was a randomized, double-blind, placebo-controlled, crossover trial in which participants took ubrogepant 100 mg or placebo for two qualifying prodrome events (10).
The co-primary endpoints for the phase 3 single-attack, headache-phase RCTs were pain freedom (PF) at 2 h post-dose and absence of most bothersome symptom (MBS) at 2 h post-dose. The comparisons between ubrogepant (50 or 100 mg, and 50 mg pooled across the two phase 3 trials) and placebo were analyzed using last observation carried forward (LOCF) and are reported as odds ratios (ORs) and 95% confidence intervals (CIs) based on logistic regression with treatment group, historical triptan response, use of medication for migraine prevention, baseline headache severity, sex, and treatment group by sex interaction as explanatory variables. A pooled analysis of ubrogepant (50 and 100 mg) was also performed across the two phase 3 trials using observed cases (OCs). This pooled analysis evaluated PF and absence of MBS over 48 h post-dose.
The primary endpoint for the phase 3 prodrome crossover RCT was absence of headache of moderate or severe intensity within 24 h post-dose. Analysis consisted of a generalized linear mixed model (GLMM with an identity link function) using observed cases (with treatment group and treatment period as categorical fixed effects).
Atogepant
The atogepant phase 3 preventive episodic migraine RCTs consisted of 12-week treatment periods and enrolled adults experiencing 4–14 migraine days and <15 headache days per month. One episodic migraine RCT included participants who were previously failed by 2–4 classes of conventional oral migraine preventives. The atogepant chronic migraine RCT enrolled adults experiencing ≥15 headache days per month, including ≥8 migraine days. Atogepant 10 mg (episodic migraine), 30 mg (episodic migraine), and 60 mg (episodic migraine and chronic migraine) once daily and 30 mg twice daily (chronic migraine) were the doses of interest for these analyses. Pooled analyses only include data from episodic migraine trials.
The primary endpoint for all four atogepant RCTs was change from baseline in mean monthly migraine days (MMDs) across the 12-week treatment period. These data are presented as least-squares mean difference (LSMD) and 95% CI between atogepant and placebo in males and females. Comparisons between each atogepant group and placebo were made using a mixed-effects model for repeated measures (MMRM) of change from baseline.
Exposure-response analyses
Post hoc exposure-response analyses were conducted with the total population using pooled ubrogepant 25, 50, and 100 mg data from phase 3 single-attack RCTs and atogepant 10, 30 and 60 mg data from phase 3 episodic migraine and chronic migraine RCTs. These analyses assessed the relationship between plasma concentrations at 2 h for ubrogepant and average plasma concentrations (Cavg) for atogepant and treatment response on PF at 2 h and change from baseline in mean MMDs across 12-week treatment period, respectively.
Ubrogepant and atogepant plasma concentrations were analyzed continuously and by quartiles. For ubrogepant, logistic regression modeling for PF at 2 h using observed concentrations at 2 h and participant sex as predictors was conducted. For atogepant (episodic migraine and chronic migraine separately), a linear regression model for MMDs was conducted using population pharmacokinetic (PK) modeling to predict atogepant Cavg with baseline MMDs and participant sex as predictors. Different shapes for the exposure-response analyses were tested linearly, logarithmically, and for maximum effect (Emax), and the optimal model was determined by Akaike information criterion (AIC). Pharmacokinetic analyses included only available data, so fewer participants than the primary analyses were utilized. Chi-squared tests compared the reported models for interaction between sex and exposure.
Safety analyses
Treatment-emergent adverse events were assessed for the male and female subgroups enrolled in the ubrogepant phase 2b and phase 3 single-attack, headache-phase RCTs and atogepant pooled phase 2b/3 and phase 3 episodic migraine, and phase 3 chronic migraine RCTs.
Results
Participants
In the four ubrogepant and four atogepant RCTs, approximately 10%-14% of participants were male (Online Supplemental Table S1). The baseline demographic characteristics for males and females were generally similar, aside from the expected greater height and weight in males compared with females (Table 1). In the ubrogepant RCTs, there was a greater proportion of male than female participants who were Black/African American.
Baseline demographic characteristics for ubrogepant and atogepant randomized, double-blind, placebo-controlled trials (mITT populations).

BMI, body mass index; mITT, modified intent to treat; NR, not reported; RCT, randomized double-blind placebo-controlled trial; SD, standard deviation.
Results presented for all randomized participants.

BMI, body mass index; mITT, modified intent to treat; SD, standard deviation; TF, treatment failure; RCT, randomized double-blind placebo-controlled trial.
One participant had missing race data.
Includes Native Hawaiian/Other Pacific Islander.
Ubrogepant
Acute administration of ubrogepant during headache phase
Two phase 3 single-attack RCTs (UBR-MD-01 and UBR-MD-02) evaluated single-dose efficacy of ubrogepant versus placebo, administered during the headache phase. In UBR-MD-01, the proportion of male participants experiencing PF at 2 h post-dose (co-primary endpoint) was 14.0% (6 of 43) with ubrogepant 50 mg compared with 20.4% (10 of 49) with placebo (OR [95% CI]: 0.66 [0.22, 2.01]) (Table 2). The proportion of female participants who experienced PF at 2 h was 19.8% (75 of 379) with ubrogepant 50 mg compared with 10.8% (44 of 407) with placebo (OR [95% CI]: 2.10 [1.40, 3.15]). Similar differences were seen with ubrogepant 100 mg (Table 2). Of note, the placebo rate for 2-h PF was almost twice as high in males as in females (20.4% vs 10.8%). In UBR-MD-02, the proportion of males experiencing PF at 2 h was 19.5% (8 of 41) with ubrogepant 50 mg compared with 13.0% (7 of 54) with placebo (OR [95% CI]: 1.54 [0.50, 4.74]). The proportion of females experiencing PF at 2 h was 22.0% (93 of 423) with ubrogepant 50 mg compared with 14.4% (58 of 402) with placebo (OR [95% CI]: 1.63 [1.13, 2.34]). The treatment-by subgroup interaction did not show a difference between males and females for rates of PF in UBR-MD-01 (P = 0.1192) or UBR-MD-02 (P = 0.3247) (Table 2).
Ubrogepant versus placebo by sex subgroups for phase 3 acute treatment trials (UBR-MD-01, UBR-MD-02) and phase 3 acute treatment during the prodrome trial (3110-304-002).

CI, confidence interval; MBS, most bothersome symptom; mITT, modified intent to treat; OR, odds ratio; RCT, randomized double-blind placebo-controlled trial.
Analysis conducted using last observation carried forward.
Analysis conducted using observed cases.
Absence of MBS at 2 h (co-primary endpoint) was achieved by 40.5% (17 of 42) and 42.9% (21 of 49) of male participants receiving ubrogepant 50 mg and placebo, respectively, in UBR-MD-01 and by 26.8% (11 of 41) and 24.1% (13 of 54) of male participants in UBR-MR-02 (Table 2). For female participants, absence of MBS at 2 h was achieved by 38.4% (145 of 378) and 25.9% (105 of 405) of those receiving ubrogepant 50 mg and placebo, respectively, in UBR-MD-01 and 40.0% (169 of 422) and 27.9% (112 of 402) of those receiving ubrogepant 50 mg and placebo in UBR-MD-02. Similarly, no signal of treatment-by-subgroup interaction was observed for absence of MBS at 2 h.
Pooled data (UBR-MD-01 and UBR-MD-02) for all observed cases (OC) in female and male participants treated with ubrogepant (50 mg and 100 mg) and placebo were used to evaluate PF (Figure 1A) and absence of MBS (Figure 1B) up to 48 h post-dose. At 2 h post-dose, responses between male and female ubrogepant-treated participants were similar for PF (19.4% [26 of 134] vs 21.1% [237 of 1122]) and absence of MBS (35.1% [47 of 134] vs 39.0% [437 of 1120]). The time course of PF and absence of MBS over 48 h also showed similar response rates between ubrogepant-treated males and females with a treatment effect vs placebo at 4 h post-dose for PF (nominal P = 0.0094) and for absence of MBS (nominal P = 0.0012) in the male subgroup. The treatment-by-subgroup interaction using pooled ubrogepant (50 mg dose group) data (LOCF) did not show a difference between males and females for rates of PF (P = 0.1466) or rates of MBS (P = 0.0984)

Efficacy of pooled ubrogepant 50 mg and 100 mg vs placebo up to 48 h post-dose when administered during the headache phase (observed cases) (pooled UBR-MD-01/UBR-MD-02; mITT populations). Data after the 2-h time point include data from participants who took the optional second dose or rescue medication. aP < 0.05 ubrogepant vs placebo (males). bP < 0.001 ubrogepant vs placebo (males). cP < 0.05 ubrogepant vs placebo (females). dP < 0.001 ubrogepant vs placebo (females). mITT, modified intent-to-treat.
In the small phase 2b single-attack RCT (P006) (Online Supplemental Table S2), PF at 2 h was experienced by similar proportions of males and females treated with ubrogepant 50 mg administered during the headache phase with clear difference from placebo, while for 100 mg the differences were variable likely owing to small sample size but reflecting similar trends to other studies. Results for absence of phonophobia, photophobia, and nausea were either comparable across sexes or slightly favored males.
Acute administration of ubrogepant during prodrome (premonitory) phase
Another phase 3 trial (3110-304-002) evaluated ubrogepant 100 mg administered during the prodrome phase. Absence of headache of moderate or severe intensity within 24 h post-dose (primary endpoint) was experienced by 30.9% (17 of 55) of ubrogepant-treated qualifying prodromal events compared with 20.0% (11 of 55) of placebo-treated events in males (OR [95% CI]: 1.79 [0.86, 3.73]) and 47.7% (173 of 363) vs 29.9% (110 of 368) of events in females (OR [95% CI]: 2.16 [1.65, 2.83]) (Table 2). Absence of headache of moderate or severe intensity within 48 h post-dose (secondary endpoint) showed a treatment effect in both male (OR [95% CI]: 2.24 [1.06, 4.73]) and female (OR [95% CI]: 2.15 [1.62, 2.87]) subgroups.
Atogepant
In the pooled analyses of the modified intent-to-treat (mITT) populations of the phase 2b/3 and phase 3 atogepant RCTs conducted in episodic migraine, the placebo-corrected LSMD (95% CI) for change from baseline in mean MMDs across the 12-week treatment period (primary endpoint) with atogepant 60 mg once daily was −1.95 (−3.14, −0.75) for males and −1.13 (−1.59, −0.67) for females (Figure 2A). Similar results were observed for the pooled atogepant 10 and 30 mg once-daily dose groups. Results from the individual atogepant trials are reported in Online Supplemental Figure S1.
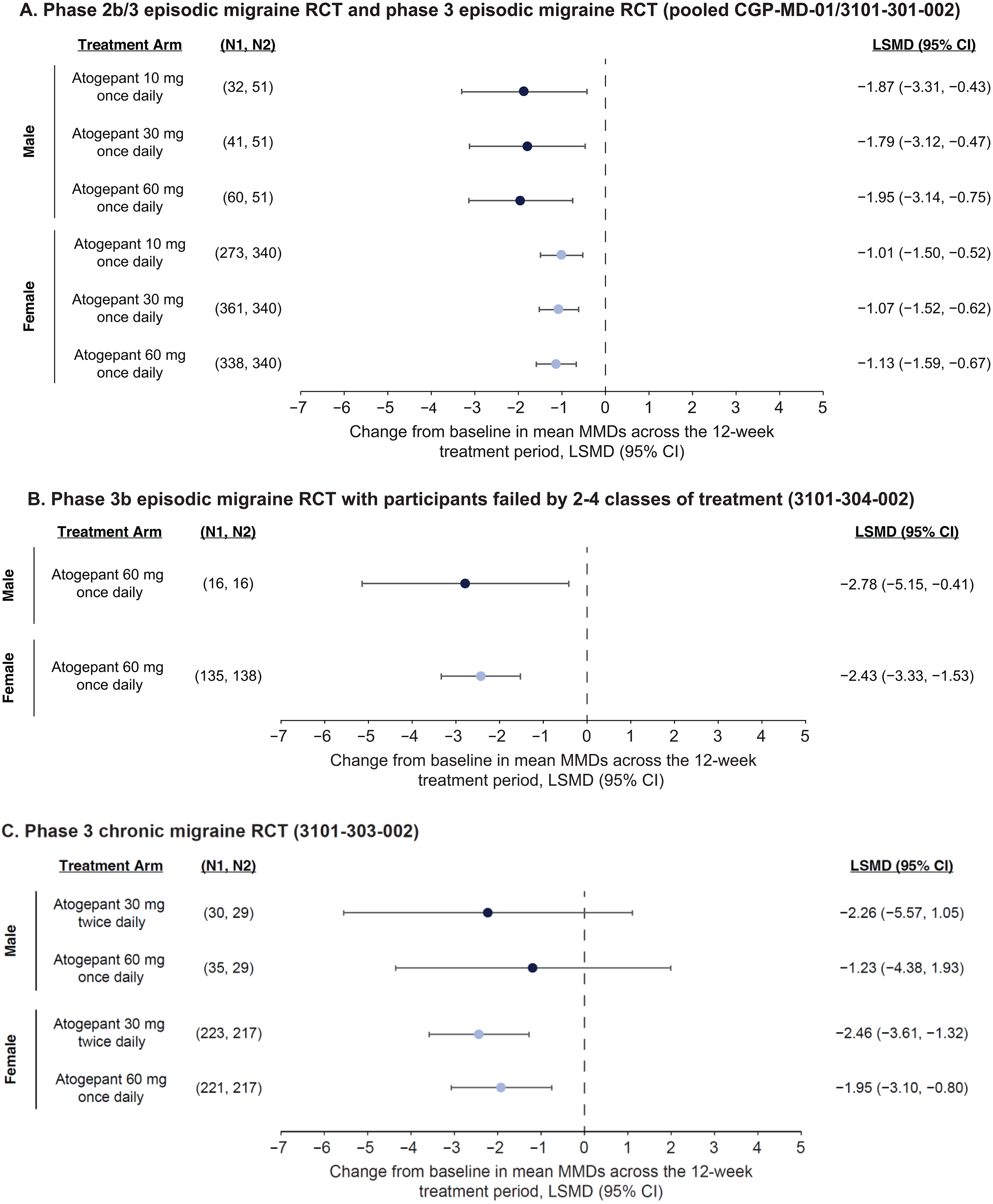
Differences in mean MMD change from baseline across 12 weeks of treatment with atogepant (mITT populations). N1, Treatment; N2, Placebo. CI, confidence interval; LSMD, least-squares mean difference; mITT, modified intent-to-treat; MMD, monthly migraine day; RCT, randomized double-blind controlled trial.
In the phase 3b episodic migraine RCT that included participants who were previously failed by prior treatment, placebo-corrected LSMD (95% CI) for change from baseline in mean MMDs across 12 weeks was −2.78 (−5.15, −0.41) for males and −2.43 (−3.33, −1.53) for females (Figure 2B).
In the phase 3 chronic migraine RCT, LSMD (95% CI) for change from baseline in mean MMDs across the 12-week treatment period was similar in females and males for atogepant 30 mg twice daily; for males it was −2.26 [−5.57, 1.05]) and for females −2.46 [−3.61, −1.32]; with atogepant 60 mg once daily it was −1.23 [−4.38, 1.93] for males and −1.95 [−3.10, −0.80] for females (Figure 2C).
Exposure-response analyses
The exposure-response analyses from pooled phase 3 single-attack RCTs showed similar rates of PF at 2 h post-dose for males and females with the highest concentration of ubrogepant (Figure 3A). In the highest ubrogepant concentration subgroup, 24.5% (73 of 310) of females achieved PF at 2 h compared with 25.0% (9 of 36) of males.

Exposure-response analyses. RCT, randomized double-blind placebo-controlled trial.
Results from three pooled atogepant RCTs in episodic migraine showed males and females had similar atogepant Cavg relative to change in MMDs (Figure 3B). Similar results were seen in the analysis of atogepant Cavg and change in MMDs for the atogepant chronic migraine trails (Figure 3C).
Pharmacokinetic parameters for ubrogepant and atogepant trended lower for males vs females (Online Supplemental Figure S2); however, all ratios were within bioequivalence criteria (0.8–1.25).
Safety/tolerability
In the phase 2b and phase 3 single-attack, headache-phase RCTs, the percentages of participants treated with ubrogepant administered during the headache phase experiencing treatment-emergent adverse events (TEAEs) were similar between males and females (Online Supplemental Table S3). Nausea was the most common adverse event for ubrogepant 50 mg and was reported by 2.8% of males and 2.5% of females, whereas in the ubrogepant 100 mg arm, nausea was reported by 1.1% of males and 5.1% of females. In comparison, the rates of nausea with placebo were 3.9% for males and 1.8% for females.
In the pooled phase 2b/3 and phase 3 migraine RCTs, the percentages of participants treated with atogepant experiencing some TEAEs, such as nausea and constipation, were numerically higher in females than males (Online Supplemental Table S3). For example, the proportions of male and female participants reporting nausea with atogepant 60 mg once daily was 5.1% and 9.7%, respectively, compared with 1.2% (males) and 3.6% (females) with placebo. Constipation was reported in 6.1% of males and 7.8% of females with atogepant 60 mg compared to 0% (males) and 2.3% (females) with placebo.
Discussion
It has been suggested that the available data do not support the effectiveness of CGRP receptor antagonists (ubrogepant, rimegepant, and zavegepant) for the acute treatment of migraine in males (18). The available data provide evidence that the CGRP receptor antagonists ubrogepant and atogepant show similar efficacy in the acute and preventive treatment of migraine, respectively, in males and females. The pooled analysis of ubrogepant (50 mg and 100 mg) RCTs found comparable responder rates in males and females for the co-primary endpoints of PF (19.4% vs 21.1%) and absence of MBS (35.1% vs 39.0%) at 2 h post-dose. Similarly, data up to 48 h post-dose demonstrated comparable response rates in males and females over time. There is an observed difference at 4 h post-dose for PF and for absence of MBS between treatment and placebo in the male subgroup. Efficacy of atogepant for the preventive treatment of migraine in males was also demonstrated. LSMDs in mean change from baseline in MMDs across 12 weeks of double-blind treatment were generally similar for both male and female subgroups in the pooled phase 2b/3 and phase 3 episodic migraine trial data, as well as the phase 3b trial in participants who were failed by 2–4 classes of oral migraine preventive treatments.
Acute treatment of ubrogepant has also been evaluated in a phase 3 crossover RCT where ubrogepant was administered during the prodrome (premonitory) phase, as opposed to during the headache phase, in individuals with migraine who were confident they could identify prodromal symptoms reliably followed by headache. Analysis of ubrogepant administered during the prodrome found similar odds ratios in males and females for the efficacy endpoints of absence of headache of moderate or severe intensity within 24 and 48 h post-dose.
Due to the lower prevalence of migraine in males compared with females, the sample size of males in ubrogepant and atogepant clinical trials was small at 10%-14% of the overall population. None of the ubrogepant or atogepant phase 3 RCTs were designed or adequately powered to evaluate treatment differences within or between sex subgroups. Due to the smaller sample sizes for males, variability in response rates in the active and placebo arms was greater. Wider confidence intervals allow for a greater likelihood of observing a placebo-corrected treatment difference in the opposite direction (e.g., placebo response greater than treatment response) by chance. A markedly higher placebo response rate (compared with active treatment) was noted in males only in ubrogepant trial UBR-MD-01. In UBR-MD-01, the placebo effect in males was 9.6 and 17.0 percentage points higher compared with females for PF and absence of MBS, respectively, at 2 h post-dose, resulting in a negative treatment difference for both ubrogepant 50 mg and 100 mg in males. However, the active arm response rates for ubrogepant 50 mg and 100 mg in males were similar to females. This higher UBR-MD-01 placebo response in males would also be evident in the ubrogepant 50 mg pooled analyses and was most pronounced until the 2 h post-dose timepoint but less so across the rest of the time course of PF and absence of MBS over 48 h.
For the other seven placebo-controlled trials in the ubrogepant and atogepant development programs, favorable placebo-corrected treatment differences were observed in both males and females for all approved doses.
A notable observation is that, despite a similar mechanism of action (e.g., CGRP receptor antagonism), a treatment effect (95% CI that excluded 0) was achieved in the male subgroup for most preventive treatment trials, but results for acute treatment trials were more variable. One explanation for this disparity may be the differences in trial designs between single-attack, acute migraine RCTs and 12-week preventive RCTs. Evaluation of efficacy as responder rates (yes/no) at individual timepoints (e.g., at 2 h post-dose) for a single migraine attack may increase the potential for variability in the results, a concern that is compounded when evaluating small sample sizes. Conversely, evaluating efficacy over a longer duration of time and across multiple migraine attacks with change from baseline endpoints, as in the 12-week preventive treatment trials, may attenuate the overall impact of variability.
Findings from nonclinical models have suggested that the function of CGRP differs in male and female rodents (19). Some evidence also suggests that CGRP receptor antagonists may function differently in male and female rats (20). However, results of population PK analyses of both ubrogepant and atogepant suggest no clinically relevant difference in their PK between male and female patients with migraine. Further, the exposure-response analyses from pooled phase 3 single-attack RCTs of ubrogepant showed no remarkable differences in response rates between males and females at the highest ubrogepant concentrations. Similarly, the relationship between average plasma concentrations of atogepant and change from baseline in MMDs was comparable between males and females. Overall, these data suggest males and females respond similarly at different levels of exposure to ubrogepant and atogepant.
Limitations
None of the four ubrogepant or four atogepant RCTs were powered to detect differences between active treatment and placebo in the smaller male populations. Additional RCTs that are sufficiently powered to evaluate acute treatment within the male population would be needed to make any definitive statements on placebo-corrected effect in males. Despite the potential limitations of these analyses, the data presented here provide a comprehensive comparison between males and females of acute and preventive migraine treatment with CGRP receptor antagonists. The strengths of these analyses include the use of RCT data for prespecified primary and secondary endpoints across multiple studies with different migraine population subsets, doses, and treatment timing scenarios; and the integration of exposure and response data for females and males.
Conclusion
Overall, these data show a similar active treatment response of the small-molecule CGRP receptor antagonists ubrogepant and atogepant in both males and females for the acute and preventive treatment of migraine.
Clinical implications
Clinical data from four ubrogepant and four atogepant randomized, double-blind, placebo-controlled trials showed male participants experienced migraine symptom improvement.
In males, ubrogepant demonstrated efficacy for the acute treatment of migraine when administered during headache and prodrome phases.
Preventive treatment with atogepant reduced monthly migraine days in males.
These individual trials were not powered to detect differences in sex subgroups.
Supplemental Material
sj-docx-1-cep-10.1177_03331024251320610 - Supplemental material for Efficacy of ubrogepant and atogepant in males and females with migraine: A secondary analysis of randomized clinical trials
Supplemental material, sj-docx-1-cep-10.1177_03331024251320610 for Efficacy of ubrogepant and atogepant in males and females with migraine: A secondary analysis of randomized clinical trials by Peter J. Goadsby, Tim P. Jürgens, Elimor Brand-Schieber, Krisztian Nagy, Yingyi Liu, Ramesh Boinpally, Sven Stodtmann and Joel M. Trugman in Cephalalgia
Footnotes
Acknowledgments
AbbVie funded this study and contributed to the study design, the collection, analysis, and interpretation of data, and the review and approval of the final manuscript for publication. All authors had access to relevant data and participated in the drafting, review, and approval of this publication. No honoraria or payments were made for authorship. The authors would like to acknowledge Chengcheng Liu for their assistance with data analysis.
Author contributions
Data sharing statement
AbbVie is committed to responsible data sharing regarding the clinical trials we sponsor. This includes access to anonymized, individual, and trial-level data (analysis data sets), as well as other information (eg, protocols, clinical study reports, or analysis plans), as long as the trials are not part of an ongoing or planned regulatory submission. This includes requests for clinical trial data for unlicensed products and indications.
These clinical trial data can be requested by any qualified researchers who engage in rigorous, independent, scientific research, and will be provided following review and approval of a research proposal, Statistical Analysis Plan (SAP), and execution of a Data Sharing Agreement (DSA). Data requests can be submitted at any time after approval in the US and Europe and after acceptance of this manuscript for publication. The data will be accessible for 12 months, with possible extensions considered. For more information on the process or to submit a request, visit the following link: ![]() .
.
Declaration of conflicting interests
The authors declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: P.J.G. reports, over the last 36 months, grants from Kallyope; personal fees from AbbVie, Aeon Biopharma, Amgen, CoolTech LLC, Dr Reddy's, Eli Lilly and Company, eNeura, Epalex, Linpharma, Lundbeck, Man & Science, Pfizer, Sanofi, Satsuma, Shiratronics, Teva Pharmaceuticals, and Vial; personal fees for advice through Gerson Lehrman Group, Guidepoint, SAI Med Partners, and Vector Metric; fees for educational materials from CME Outfitters; publishing royalties or fees from Massachusetts Medical Society, Oxford University Press, UptoDate, and Wolters Kluwer; and a patent magnetic stimulation for headache (No. WO2016090333 A1) assigned to eNeura without fee. T.P.J. reports research grants from Novartis, personal fees from AbbVie, Allergan, Grünenthal, Hormosan, Lilly, Lundbeck, Novartis, Pfizer, Sanofi, and Teva, and publishing royalties from Elsevier. J.M.T., E.B.S., K.N., Y.L., R.B., and S.S. are employees of AbbVie and may hold AbbVie stock.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and publication of this article: This study was sponsored and funded by AbbVie. Medical writing support was provided by Dennis Stancavish and Cory Hussar of Peloton Advantage, LLC, an OPEN Health company, and was funded by AbbVie.
ORCID iDs
Supplemental material
Supplemental material for this article is available online.
Notes
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.