
Abstract
Introduction
Rodent disease models can play an indispensable role in drug development. Confirming that translationally-relevant disease mechanisms are engaged in such models is a crucial facet of this process. Accordingly, we have validated the role of calcitonin gene-related peptide signaling in a mouse model of glyceryl trinitrate-provoked migraine-like pain and a spontaneous rat model of migraine-like pain by assessing their pharmacological responsiveness to the small molecule calcitonin gene-related peptide receptor antagonist olcegepant, and the humanised monoclonal calcitonin gene-related peptide antibody ALD405.
Methods
Cutaneous sensitivity to hind paw, and periorbital mechanical stimulation were used as surrogate markers of activation of relevant pain pathways in each respective model. Separate experiments were performed to identify the time-course of treatment response to olcegepant (1 mg/kg i.p.) and ALD405 (10 mg/kg i.p.).
Results
Olcegepant and ALD405 significantly alleviated cutaneous mechanical hypersensitivity in both models compared with corresponding control treatments (saline and IgG control antibody respectively). As expected, the duration of anti-nociceptive action obtained with ALD405 was considerably longer than that associated with olcegepant. Surprisingly, in the spontaneous rat model the onset of action of ALD405 occurred within just 4 hours after administration.
Discussion
The current data clearly show that calcitonin gene-related peptide-mediated signaling is critically involved in the manifestation of cutaneous hypersensitivity in distinct rodent models of migraine-like pain and emphasise their translational relevance. Moreover, the unexpected rapidity of onset observed for ALD405 supports i) a probable site of action outside the blood-brain barrier, and ii) a potential clinical utility of specific monoclonal calcitonin gene-related peptide antibodies in the abortive treatment of migraine.
Introduction
The onset of a migraine attack is preceded by the activation of trigeminal sensory neurones and the release of several vasoactive substances from their peripheral terminals residing in the dural vasculature (1–3). One of the principal substances is the 37 amino acid peptide neurotransmitter calcitonin gene-related peptide (CGRP) which is produced by alternative splicing of the calcitonin gene (1,4–7). Accordingly, the peripheral administration of CGRP results in a delayed migraine-like headache in migraine patients (6) and produces spontaneous pain in mice that is characterised by orbital tightening (8). CGRP is also released from the central terminals of sensory neurones together with other excitatory transmitters such as glutamate and contributes to ongoing neuronal hyperexcitability within central pain circuits (2,9–11). Importantly, this is linked to the process of central sensitisation, which is a neurobiological correlate of the cutaneous allodynia that frequently accompanies migraine pain in patients (12,13).
Animal models of migraine-like pain enabling evaluation of disease-relevant behaviors sensitive to anti-migraine drugs are frequently provoked by chemical triggers such as the nitric oxide (NO) donor glyceryl trinitrate (GTN) (14–16). Whereas acute administration of GTN produces a short-lasting hyperalgesia observed as a reduction in head or hind paw thresholds to cutaneous mechanical stimulation, its repeated administration produces an additional basal hyperalgesia that can last for a number of days, likely as a consequence of additional pathophysiological mechanisms being recruited (14,15). An alternative approach to assess longer-term migraine-relevant mechanisms in rodents is via the use of congenital models (17). The spontaneous trigeminal allodynia (STA) rat is a unique inbred rat strain (18) that exhibits a persistent cutaneous cephalic mechanical hypersensitivity, which is sustained into adulthood and is sensitive to treatment with sumatriptan (19).
The back-translational utility of such models for predicting efficacy of small molecule and biological therapeutics targeting CGRP signaling mechanisms relevant to migraine within the same laboratory settings has never been tested. Moreover, the precise site(s) of action within peripheral and/or central pain circuits for CGRP receptor antagonists (20–22), and even CGRP antibody-based therapeutics efficacious in the prophylaxis of migraine, remains unresolved (23). In the current study we have addressed these issues, in part, by comparing the efficacy of the CGRP receptor antagonist olcegepant with the humanised monoclonal CGRP antibody ALD405 in GTN-treated mice and STA rats. Our data confirm that these models can play a pivotal role in migraine drug development and provide further insights into the future clinical potential of CGRP antibody therapeutics in migraine.
Materials and methods
Animals and housing
For mouse experiments, male C57Bl/6J mice (Taconic, Denmark) of 8–9 weeks age and body weight 22–30 g were used for the experiments following 7 days of acclimatisation to the animal facility. Mice were group-housed (2–8 mice) in type III 1284L IVC cages (L × W × H: 36.5 × 20.7 × 14.0 cm; Tecniplast, Italy) and allowed free access to food (Altromin 1314, GmbH & Co., Germany) and tap water. Cages were floored with sawdust (Aspen chip, Tapvei, Finland) and enriched with a red-tinted polycarbonate shelter (Molytex, Denmark), nesting material (Happi-Mats, Scanbur, Denmark) and biting sticks (Tapvei, Finland). The light cycle was 12/12, light on at 06.00 h. Humidity ranged from 45–55%. Animals were weighed every other day in relation to dosing of test compounds. In total, 107 mice were used in the study. All experiments were planned and carried out in accordance with ARRIVE guidelines under license number 2017-15-0201-01358 from the Danish Animal Experiments Inspectorate and project number P18-350 from the Department of Experimental Medicine, University of Copenhagen.
For rat experiments, adult female STA rats (n = 20 in total; body weight 226–276 g; age 14–18 weeks and 235–269 g; age 17–21 weeks for ALD405 and olcegepant experiments, respectively), derived from a single breeding pair originally sourced from the Thomas Jefferson University under license and subsequently bred in house, were used. Periorbital thresholds for this breeding pair, measured via manual von Frey filament testing, have been described previously (19). An additional group of adult female Sprague-Dawley rats (n = 10; body weight 242–278 g) were included specifically for measuring periorbital thresholds to ensure that STA rats presented with an appropriate cephalic hypersensitivity prior to pharmacological testing. All rats were group-housed (4–5 rats) in Tecniplast 1354G Eurostandard type IV polycarbonate cages (L × W × H: 60 × 38 × 20 cm; Brogaarden, Denmark) using a 12-hour light/dark cycle with lights on at 04.00 h. Two separate opaque red polycarbonate shelters (20 × 11.5 × 16 and 15 × 9 × 9 cm respectively), together with an aspen biting stick (10 × 2 × 2 cm; Tapvei, Estonia) and piece of hemp rope suspended from the cage lid were provided in each home cage for retreat and enrichment purposes. Bedding consisted of Enviro-Dri nesting material (Brogaarden, Denmark). Standard rat chow (Altromin 1314, GmbH & Co., Germany) and tap water were available ad libitum in the animals' home cage environment. Humidity ranged from 45–55%. Experiments were approved by the Danish Animal Experiments Inspectorate and performed in accordance with the relevant guidelines and regulations of approval numbers 2012-15-2934-00697 and 2014-15-0201-00256.
Compounds
The NO donor GTN (7.89 mg/ml in 96% ethanol, Cambrex Germany, distributed via the Capital Region, Hospital Pharmacy) was diluted in saline to 1 mg/ml and administered to mice at 10 mg/kg (14,15). For vehicle treatment, the same amount of ethanol was dissolved in saline. The small molecule CGRP receptor antagonist olcegepant HCl (MedChemTronica AB, Sweden) was dissolved in saline (0.1 mg/ml for mice and 1 mg/ml for rats) and administered at 1 mg/kg (19). Note that mice received 5% DMSO in saline as a vehicle control for olcegepant as the experiment originally included a fourth treatment group (compound X + GTN) where compound X was dissolved in DMSO; data for compound X are not included here as they are not relevant to the hypothesis being tested. The selective 5HT1B/1D receptor agonist sumatriptan (GSK Denmark, Imigran 12 mg/ml) was diluted in saline to 0.06 mg/ml and administered at 0.6 mg/kg to mice (14,24). The humanised monoclonal CGRP antibody (ALD405) together with the isogenic IgG control antibody and vehicle solution were kindly donated by Alder Pharmaceuticals. ALD405 and the IgG control antibodies were diluted in vehicle (1 mg/ml for mice and 10 mg/ml for rats) and dosed at 10 mg/kg as recommended by the provider. All substances were injected intraperitoneally in a volume of 10 ml/kg.
Mechanical sensitivity thresholds in GTN mice
In mice, cutaneous mechanical sensitivity was tested as described by Pradhan and colleagues (14,15) by applying a series of von Frey filaments (Ugo Basile) to the plantar surface of the left hind paw. The 50% withdrawal threshold was determined as originally defined by Chaplan et al. (25). Filaments were applied until a rapid withdrawal of the paw occurred. In the absence of such a positive response the filament was applied for 2–3 seconds, at which point it was removed. During testing the animals were kept in individual clear plexiglass cages (L × W × H: 10 × 10 × 12 cm) with a mesh floor (IILC Life Science, US) allowing access to the plantar surface of the paws. For testing of periorbital thresholds, a round plastic cup (D × H: 8 × 5.8 cm) was placed with the bottom surface facing upwards within each individual plexiglass cage. The cup served as a platform for the mouse to rest on and enabled the experimenter to access the periorbital area with von Frey filaments from above, which would otherwise have remained out of reach. To facilitate hind paw testing, mice were acclimatised to the chambers for 30–45 minutes, and for periorbital testing they were placed in the chamber set-up 1 hr prior to testing and on the 2 days preceding day 1. Furthermore, to avoid overtly sensitising the mice to the visual presentation of the filament, periorbital testing was not performed on days 3 and 7. All testing was performed between 07.30–16.00 h.
Measurement of periorbital and hind paw thresholds after mechanical stimulation in STA rats
The sensitivity of the frontal region of the head (V1 ophthalmic trigeminal dermatome) to static mechanical stimulation was measured using an electronic von Frey device fitted with a rigid plastic tip (IITC Life Science Inc, USA) as described previously (19) with testing performed between 08.00–16.00 h. The rat was gently restrained using a cotton towel and held in a prone position in the lap of the investigator with its head and neck region left exposed and unrestricted. The tip of the device was then applied with increasing force (maximum of 450 g) to the right periorbital area above the eye until the rat withdrew its head, laterally rotated its head and/or vocalised. The soft tissue around the eye was carefully avoided. The process was then repeated over the midline, the left and then the right periorbital areas above the eye. The average of the last three measurements was considered the withdrawal threshold (g). If a stimulus had to be reapplied due to inappropriate application, care was taken not to apply the probe tip to the exact same location within the region being measured. The whole procedure typically took less than 2 minutes to perform. We have previously shown that there is no difference in periorbital thresholds in female STA rats during different stages of the oestrus cycle (19).
Hind paw sensitivity to mechanical stimulation was assessed using the same electronic von Frey device as described previously (19). Briefly, the rat was gently restrained within the hand and lower arm of the investigator and the right hindleg extended and the hind paw inverted. The probe tip was then placed on the paw between the foot pads and increasing force uniformly applied (maximum of 450 g) until the rat withdrew the hind paw and/or vocalised. The average of three measurements obtained from defined parts of the hind paw excluding the footpads was considered the paw pressure withdrawal threshold (g).
Assessment of motor function in STA rats
To assess if motor function was putatively impaired by drug treatment, a simple observational analysis based on accumulated motor scores was made according to previously described methods (26,27). Immediately after threshold testing, the rat was placed onto a clean tabletop and hindlimb gait scored as (2, normal; 1, moderate hindlimb weakness/splaying; 0, complete hindlimb weakness/splaying). The ability to walk was then scored as (2, normal; 1, walks with mild deficit; 0, no attempt to walk). Next, the dorsum of the right hind paw was drawn across the edge of the table, a stimulus which elicits a lifting of the paw onto the surface of the table and scored as (normal, 2; delay of 1–2 s, 1; delay >2 s, 0). Finally, the whole-body righting reflex was assessed by placing the rat horizontally with its back on the table, which normally gives rise to an immediate, coordinated twisting of the body to an upright position (normal, 2; delay of 1–2 s, 1; delay > 2 s, 0). Each test was performed once, and the total score was summed for each rat. The complete battery of tests took less than 1 minute to perform for each rat.
Experimental protocols in GTN mice and STA rats
Description of experimental groups in GTN mouse and STA rat experiments. All drug treatments were administered i.p.

Two mice were excluded after the study was initiated (one due to a broken tooth with subsequent weight loss and one due to an adverse reaction after injection of the control IgG antibody).
SD: Sprague-Dawley.
Similarly, for the STA rats two separate experiments were performed with ALD405 and olcegepant (Table 1). In the first experiment, after periorbital and hind paw baseline thresholds had been assessed, STA rats were injected with either ALD405 (10 mg/kg, i.p., n = 10) or the isotype control IgG antibody (10 mg/kg, i.p., n = 10). Thereafter, regular threshold responses were obtained at 2, 4 and 6 h and then 1, 3, 4, 7, 8, 10, and 14 days post-treatment. After a wash-out period of 3 weeks, STA rats previously treated with the isotype control antibody were re-used to assess the comparative efficacy of olcegepant. In this second experiment after baseline periorbital and hind paw thresholds had been assessed, STA rats were injected with either olcegepant (1 mg/kg, i.p, n = 5) or vehicle (n = 5) and threshold responses measured regularly at 1, 2, 4, 6 and 24 h post-treatment. After a wash-out period of 2 days, rats were then crossed over to the corresponding treatment and the process repeated to obtain final group sizes of 10 rats for olcegepant and vehicle. To confirm that any drug effects on reflex threshold responses did not simply occur due to non-discriminative actions on efferent motor output, motor function was assessed in STA rats in each experiment.
The persons determining the threshold measures in GTN mice and STA rats were blinded to treatment in all cases.
Data handling and statistics
For both mouse and rat experiments, randomisation was performed within individual home cages to avoid any cage effects, and balanced to have approximately equal numbers of each treatment group represented in each home cage. Group sizes for the mouse experiments were based on previously published data (14). For the STA rat pharmacology experiments assessing cutaneous threshold values, group sizes were estimated as a function of the desired effect size (approximately 50% change versus corresponding vehicle treatment or with baseline control values), where we assumed a significance level of 5% and a power of 90% (30) based on a sample size of 10 per group.
Statistical analysis was performed using GraphPad Prism 7.02 (Graph Pad Software Inc., San Diego, CA, USA). Parametric assumptions were evaluated for all variables using histograms, descriptive statistics and the Shapiro-Wilk test for normality. For the mouse GTN experiments, to obtain normally distributed data with equal variances, the paw threshold data were transformed by taking the square root of individual 50% threshold values. For the STA rat experiments, periorbital and hind paw threshold data were normally distributed. Thereafter, two-way repeated measure (RM) ANOVA and Bonferroni correction for multiple comparisons between test groups was performed. Data are presented as mean ± standard error of mean (SEM) unless specified otherwise. Comparisons of the accumulated motor scores between treatment groups in STA rats were made using an unpaired t test with Welch's correction. p < 0.05 was considered statistically significant.
Results
Olcegepant prevents acute hyperalgesia, but not basal hyperalgesia in GTN mice
Basal mechanical thresholds and acute responses to GTN provocation are shown in Figure 1(a) and (b), respectively. Overall, ANOVA indicated a significant effect for the different treatment groups (F(2,32) = 49.86, p < 0.001). More specifically, post hoc analysis revealed that GTN induced a prominent acute hyperalgesia response 2 h after administration compared with vehicle treatment, and that this response was significantly inhibited by olcegepant on every test day (Figure 1(b)). Interestingly, the effect of olcegepant was less pronounced on days 7 and 9, albeit it was still significantly different from the vehicle control group. Furthermore, GTN-treated mice developed a progressive basal hyperalgesia (F(2,32) = 37.19, p < 0.001). This was evident already after a single GTN administration and could not be prevented by olcegepant (Figure 1(a)).
Olcegepant prevents GTN-induced acute hyperalgesia in mice. Following baseline measurements of 50% paw withdrawal thresholds GTN (10 mg/kg, i.p.) or vehicle was given 15 minutes after olcegepant (1 mg/kg, i.p.) or vehicle on every test day (1, 3, 5, 7 and 9). The acute GTN response was measured 2 h after drug administration. GTN induced (a) basal and (b) acute hyperalgesia and olcegepant clearly prevented the expression of acute hyperalgesia on all test days. Raw data was square root transformed and represented as mean ± SEM.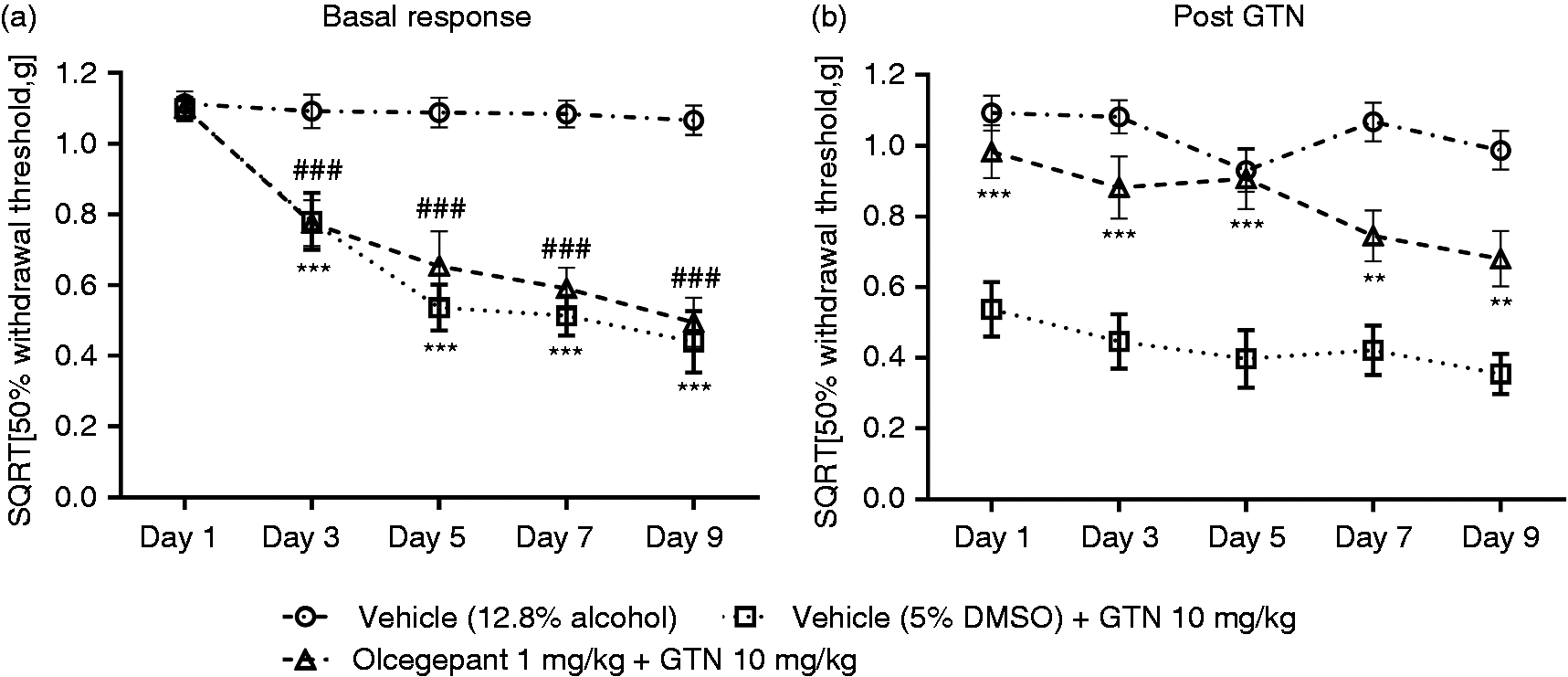
ALD405 prevents both acute and basal hyperalgesia in GTN mice
Mouse responses to ALD405 and GTN treatments are depicted in Figure 2(a) and (b). Following administration of ALD405 and its isotype IgG control on day 0, all three groups had 50% withdrawal thresholds that were similar to naïve mice prior to GTN administration on day 1 (Figure 2(b)). ALD405 inhibited the acute response to GTN (F(3,41) = 9.48, p < 0.001) on day 1, 3 and 5 (Figure 2(d)). By day 7, the effect of ALD405 had declined and the ALD405 + GTN treatment group was not significantly different from the IgG + GTN treatment group at this time (p > 0.99). However, at day 9 the effect was again significant (p = 0.013). By day 13, the responses to GTN in the ALD405 and IgG control groups were again at the same level (p = 0.59). The development of basal hyperalgesia was completely inhibited by ALD405 (F(3,42) = 12.13, p < 0.001) throughout the test protocol (Figure 2(a)) compared to IgG controls. Throughout both of the GTN mouse experiments assessing efficacy of ALD405 and olcegepant, no adverse effects to test compounds were observed with the exception of one mouse, as described in Table 1. All mice appeared to exhibit normal body tonus, locomotor activity and showed no signs of body weight loss throughout the duration of the experiment.
ALD405 prevents GTN-induced acute and basal hyperalgesia in mice. ALD405 (10 mg/kg, i.p.) or isotype control antibody (10 mg/kg, i.p.) was given on day 0. Following baseline measurements of 50% paw withdrawal thresholds GTN (10 mg/kg, i.p.) or vehicle was administered on every test day (1, 3, 5, 7, 9 and 13). The acute GTN response was measured 2 h after injection. No GTN indicates a period during which GTN was not administered to mice. GTN induced (a) basal and (b) acute hyperalgesia. ALD405 completely prevented the expression of basal hyperalgesia for at least 9 days and the presence of acute hyperalgesia for 5 days. Raw data was square root transformed and represented as mean ± SEM.
Finally, to confirm that GTN administration in mice also sensitises trigeminal pain circuits in a manner that is consistent with sensitivity to migraine-specific drugs, we assessed the effect of sumatriptan on GTN-induced periorbital hypersensitivity (Figure 3). There was an overall difference between the test groups (F(2,21) = 47.03), p < 0.001), as revealed in the post hoc test showing a significant effect of GTN as compared to vehicle on every test day (p < 0.001) and a significant sumatriptan reversal of the GTN-induced hyperalgesia (p < 0.004). Accordingly, our data align with that reported by other groups, which have shown that the GTN mouse model presents with trigeminal and somatic hyperalgesia, both of which are responsive to anti-migraine drugs (14,31).
GTN induces periorbital hyperalgesia that can be reversed by treatment with sumatriptan. GTN (10 mg/kg, i.p.) or vehicle (12.8% alcohol) were administered every second day (1, 3, 5, 7 and 9). Sumatriptan (0.6 mg/kg, i.p.) or vehicle were administered 45 min prior to GTN on test days 1, 3, 5, 7 and 9. Note that periorbital sensitivity to mechanical stimulation measured 2 hrs after GTN administration was only measured on tests day 1, 5 and 9. GTN robustly induced acute hyperalgesia and sumatriptan prevented this on each test day. Raw data was square root transformed and represented as mean ± SEM.
ALD405 and olcegepant selectively reverse cephalic hypersensitivity in STA rats
Next, two separate experiments were performed in adult female STA rats with ALD405 and olcegepant to compare onset and duration of action of a CGRP antibody versus a small molecule CGRP receptor antagonist against cephalic hypersensitivity. As expected, STA rats presented with low periorbital thresholds to cutaneous mechanical stimulation (98.2 ± 3.6 g, n = 20). In contrast, the periorbital threshold to mechanical stimulation (242.6 ± 17.0 g, n = 10) was significantly higher in a control group of adult female Sprague-Dawley rats (p < 0.001, unpaired t-test). Figure 4(a) shows that the periorbital threshold to mechanical stimulation in STA rats was increased significantly by a single i.p. injection of 10 mg/kg ALD405 (treatment × time interaction F(10,180) = 4.929, p < 0.001). Post-treatment analysis revealed that the effect of ALD405 treatment was already apparent at 4 h after injection compared with the isotype IgG control (p < 0.05). Maximal efficacy of ALD405 occurred from 6 h through to 4 days after administration (all p < 0.001) before gradually diminishing thereafter. In contrast, ALD405 had no effect on hind paw thresholds to mechanical stimulation (F(9,180) = 0.8504, p = 0.5708), (Figure 4(c)).
ALD405 reverses cephalic hypersensitivity in STA rats with a prolonged duration of action compared with olcegepant. Cutaneous periorbital and hind paw thresholds to mechanical stimulation were measured at baseline. Effects of the CGRP antibody ALD405 (10 mg/kg, i.p., n = 10) or isotype control antibody (10 mg/kg, i.p., n = 10) at the time indicated on (a) periorbital thresholds and (c) hind paw thresholds. Effects of the small molecule CGRP receptor antagonist olcegepant (1 mg/kg, i.p., n = 10) or vehicle (n = 10) at the times indicated on (b) periorbital thresholds (d) hind paw thresholds. Note that olcegepant reversed cephalic hypersensitivity more rapidly in STA rats than ALD405. However, ALD405 had a far greater duration of action than olcegepant. Data represent mean ± SEM.
After a prolonged 3-week period of wash-out we administered olcegepant (1 mg/kg, i.p.) to STA rats previously administered the isotype IgG control antibody. Figure 4(b) shows that cephalic hypersensitivity in these STA rats was significantly diminished by acute administration of olcegepant (treatment × time interaction F(5,45) = 18.14, p < 0.001). Olcegepant had a rapid onset of action, as observed by the increase in periorbital threshold to mechanical stimulation at 1 h post-treatment (p < 0.001 vs. vehicle), and this effect was maintained for at least a further 3 h, before gradually dissipating at 6 h after administration. Note that this effect of olcegepant had completely resolved at the 24 h timepoint, in marked contrast to ALD405. However, similarly to ALD405, olcegepant had no effect on hind paw threshold responses to mechanical stimulation (F(4,36) = 1.803, p = 0.1497), (Figure 4(d)).
Finally, to confirm that the efficacy of ALD405 and olcegepant on reflex nociceptive behaviours in STA rats was mediated specifically via trigeminal pain circuits and not indiscriminate actions on motor behaviour, we assessed the accumulated motor scores in each experiment. Accordingly, 2 h after administration of olcegepant and 24 h after administration of ALD405, the accumulated motor scores were 8, 8, 8 (median, 25th percentile, 75th percentile) after each treatment. These values were unchanged in the respective vehicle-treated groups. The sensitivity of the assay has been confirmed previously in rats via administration of the GABAA receptor positive allosteric modulator midazolam (1 mg/kg, s.c.), wherein accumulated motor scores are reduced to 3.5, 3.0, 4.75 compared with vehicle treatment (8, 7.25, 8; p = 0.0136, Wilcoxon matched pairs signed rank test), (unpublished data). Thus, neither olcegepant nor ALD405 affected motor function in STA rats.
Discussion
We have used two distinct therapeutic strategies that target either CGRP peptide or the CGRP receptor to unequivocally show that CGRP-mediated signalling mechanisms play a key role in the manifestation of cutaneous hyperalgesia in rodent models of migraine-like pain. This is the first time that the efficacy of a humanised monoclonal antibody has been compared directly with a small molecule receptor antagonist in such a setting. Importantly, it underscores the back-translational utility of these models in relation to understanding basic migraine mechanisms, and their value in the future development of migraine therapeutics. Moreover, the unexpectedly fast onset of action achieved with ALD405 in STA rats indicates that i) CGRP related antibody therapeutics do not appear to require access into the CNS to reduce migraine-like pain and ii) may have some clinical utility in the acute treatment of migraine.
GTN mice
The systemic administration of the NO donor GTN is a widely used method to model facets of experimental migraine in rodents and humans. Both healthy volunteers and migraine patients experience a mild, transient headache immediately after GTN provocation (32–34). Although GTN on its own appears to precipitate a delayed onset migraine attack only in migraineurs, when combined with the carbonic anhydrase inhibitor acetazolamide, over sufficient time a migraine-like headache can occur even in normal subjects (35). Correspondingly, in rodents, the acute systemic administration of GTN has been reported by various groups to i) produce a short-lasting cutaneous mechanical and/or thermal hypersensitivity within somatic and trigeminal pain circuits (24,36), ii) increase the firing rate of first order neurones within the trigeminal cervical complex (37) and iii) result in activation of several brain regions relevant to migraine (24,38,39). Thereafter, with repeated administration of GTN, a basal hyperalgesia emerges most likely as a consequence of central hyperexcitability mechanisms reflective of central sensitisation being engaged (14,15). We confirmed the presence of both peripheral and cephalic acute and basal cutaneous mechanical hyperalgesia in GTN-injected mice in our experiments, thereby underscoring the reproducibility of the model when performed under laboratory conditions that can differ in relation to, for example, the provider/formulation of GTN, investigator, animal vendor, sex, and strain of test subject. This is important because reproducibility of pain models and predictivity of pharmacological outcome has been suggested to be a possible limitation in pain drug development (40,41).
Olcegepant clearly blocked the functional expression of acute GTN-mediated hyperalgesia in mice, confirming other observations obtained in rats that CGRP signalling mechanisms contribute to the manifestation of this process (38,42). However, these studies utilised single injection or infusion of GTN, in contrast to our experiments. Accordingly, we observed that the initial robust efficacy obtained with olcegepant diminished over time, and clearly failed to prevent the development of the basal, longer-lasting hyperalgesia. The simplest explanation for the latter finding likely relates to the relatively short plasma half-life of olcegepant in rodents (K Rudolfs, Boehringer Ingelheim Pharma, personal communication). On the other hand, the tachyphylaxis seen at Day 9 after the final injection of olcegepant might be explained by a diminished contribution of peripheral CGRP-mediated signalling in trigeminal afferent drive to the CNS with increasing disease chronification (14,15,22), albeit chronic administration of GTN in rats has been shown to upregulate CGRP gene expression in areas relevant to migraine pain (43,44). ALD405 also had a pronounced effect on acute GTN-mediated hyperalgesia, but in contrast to olcegepant it prevented the development of basal hyperalgesia. These observations are consistent with other studies in which ALD405 (30 mg/kg, i.p.) has been shown to diminish light sensitivity and orbital tightening in response to systemically administered CGRP (8). Similarly, the humanised CGRP antibody TEV-48125 inhibits cephalic and hind paw tactile thresholds in response to bright light stress and the NO donor sodium nitroprusside (45). Taken together, these findings support the back-translational utility of this animal model in migraine drug discovery (17).
STA rats
A possible limitation associated with the use of provocation agents such as GTN in long-term behavioural studies in rodents is that the basal hyperalgesia resolves a number of days after the final injection (14). However, this issue can be circumvented via the use of spontaneously-occurring models of migraine-like pain. We have recently shown that STA rats display a sex-independent, persistent cutaneous mechanical hypersensitivity exclusively within trigeminal dermatomes that can be reversed by clinically effective anti-migraine drugs such as sumatriptan (19). This feature allowed us to assess the efficacy of ALD405 in these rats against established pathophysiological changes in a manner congruent to the clinical assessment of such therapeutics in patients with chronic migraine. Accordingly, ALD405 was shown to effectively reverse cephalic hypersensitivity in female STA rats within hours after administration. Olcegepant also diminished cephalic hypersensitivity in STA rats, in agreement with our previous observations and other rat models of migraine-like pain triggered by NO donors (19,46). However, whereas olcegepant efficacy diminished within hours, ALD405 continued to be effective for at least 4 days after administration, consistent with its far greater plasma half-life in rats, which is in the range of 7 days.
The issue of whether drugs targeting CGRP signaling mechanisms related to migraine act within the peripheral and/or central nervous system remains unresolved (9,11,47). Crucially, the exquisite selectivity and target affinity of CGRP antibodies implies that even a fractional exposure within the central nervous system might be sufficient to contribute to their efficacy. However, a recent pharmacokinetic study performed in rats and monkeys has reported that the systemic administration of five distinct monoclonal antibodies is associated with slow uptake into cerebrospinal fluid, taking 24–72 h to reach steady-state cerebrospinal fluid to serum ratios of just 0.1–0.2% (48). Taken together with the rapid onset of action achieved with ALD405 in STA rats, we would surmise that for CGRP antibodies at least their mechanism of action resides outside the central nervous system.
Conclusions
Our results with olcegepant and ALD405 support the translational utility of two distinct rodent models of chronic migraine-like pain used in preclinical drug discovery. The rapid onset of action obtained with ALD405 in STA rats was surprising for an antibody therapeutic. Our findings argue strongly that drugs targeting CGRP peptide or its receptor have a peripheral site of action. It remains to be explored if CGRP antibodies can be used in the acute treatment of migraine.
Footnotes
Article highlights
Cutaneous hyperalgesia relevant to migraine in GTN mice and STA rats was reversed by the CGRP receptor antagonist olcegepant and the humanised monoclonal antibody ALD405.
ALD405 had a far longer duration of action than olcegepant in both models.
ALD405 had a rapid onset of action in STA rats, supporting a peripheral site of action on CGRP-mediated signalling.
Author contributions
GM, SLC, DMK and JO designed the study. GM, SLTC and SP performed all experiments and data analysis. GM and SLTC wrote the manuscript. All authors read and approved the final manuscript.
Declaration of conflicting interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: Financial support was provided by Candys Foundation.