
Abstract
Background
ATP1A2 has been identified as the genetic cause of familial hemiplegic migraine type 2. Over 80 ATP1A2 mutations have been reported, but no data from Chinese family studies has been included. Here, we report the first familial hemiplegic migraine type 2 Chinese family with a novel missense mutation.
Methods
Clinical manifestations in the family were recorded. Blood samples from patients and the unaffected members were collected for whole-exome sequencing to identify the pathogenic mutation. Seven online softwares (SIFT, PolyPhen-2, PROVEAN, PANTHER, MutationTaster2, MutationAssessor and PMut) were used for predicting the pathogenic potential of the mutation. PredictProtein, Jpred 4 and PyMOL were used to analyze structural changes of the protein. The mutation function was further tested by Methylthiazolyldiphenyl-tetrazolium bromide (MTT) assay.
Results
All patients in the family had typical hemiplegic migraine attacks. Co-segregation of the mutation with the migraine phenotype in four generations, with 10 patients, was completed. The identified novel mutation, G762S in ATP1A2, exhibited the disease-causing feature by all the predictive softwares. The mutation impaired the local structure of the protein and decreased cell viability.
Conclusion
G762S in ATP1A2 is a novel pathogenic mutation identified in a Chinese family with familial hemiplegic migraine, which causes loss of function by changing the protein structure of the Na+/K+-ATPase α2 subunit.
Introduction
Familial hemiplegic migraine (FHM) is a rare type of migraine with aura. Transient hemiplegia during the aura is the typical feature (1). FHM is an autosomal dominant disorder for which three pathogenic genes have been identified: CACNA1A, ATP1A2 and SCN1A (1). Besides FHM, these three genes have also been identified in some patients with sporadic hemiplegic migraine (SHM) (2).
ATP1A2 encodes the α2 isoform of the sodium/potassium-ATPase (Na+/K+-ATPase, NKA) catalytic subunit (3). The catalytic subunit of NKA utilizes the free energy of ATP hydrolysis to export three sodium ions and import two potassium ions per ATP molecule, which is responsible for the maintenance of electrochemical gradients of both ions across the plasma membrane (4). These gradients are essential for the sustainment of resting potential and the electrical excitability of neuronal cells and muscles (4). The α2 subunit protein, which consists of 10 transmembrane (TM) helices, is especially highly expressed in astrocytes in the central nervous system (3). Among these TMs, TM5 is critical for cation combination and transport. The TM4-5 loop on cytoplasmic side preserves the longest amino acid sequence (433 residues), which contains the phosphorylation domain (P domain), the nucleotide binding domain (N domain), and the part between the TM region and P domain (5,6) (Figure 1(a)). The sequence of the TM4-5 loop is highly conserved, and links two important functional domains and the channel for cation transport, TM5. However, it possesses the most missense mutations of ATP1A2, with 37 reported mutations, which often induce NKA dysfunction (7).
Location of G762S in the ATP1A2 protein topology. As a novel mutation, G762S is depicted in red. Mutations tested in former functional studies are marked in purple.
Mutations of ATP1A2 have been considered to cause dysfunction in the α2 subunit of NKA, inducing brain hyperexcitability, which is followed by cortical spreading depression (CSD). CSD is considered to be the essential part to induce the process of migraine aura. Over 70% of the reported mutations have been tested in functional studies in vitro, such as ouabain survival assays, biochemical assays, and electrophysiological assays. These mutations were shown to be responsible for hemiplegic aura in FHM or SHM patients (Figure 1) (7). The W887R knock-in mouse model has been generated and confirmed in vivo that W887R decreased the threshold of inducing CSD and increased the velocity of propagation (8).
Here, we found a family with FHM, recorded their clinical phenotypes, and screened their genome sequence. A novel mutation located in the TM4-5 loop of ATP1A2, G762S, was found. Then, we analyzed the pathogenicity of G762S using mutation-predicting softwares. We also used Jpred4, PredictProtein and PyMOL to analyze the changes in protein structure. Furthermore, we tested the prediction results by Methylthiazolyldiphenyl-tetrazolium bromide (MTT) assay in the lab.
Methods
Clinical data collection
Two headache specialists interviewed the family, collected clinical data of each family member and made diagnoses according to the International Classification of Headache Disorders 3rd edition (ICHD-3) beta criteria (9). Data of 16 consanguineous individuals and 10 spouses were collected, including 10 affected members and 16 unaffected ones (Figure 2(a)). Three of the affected members (I-1, II-1, III-6) were deceased, and their clinical histories were recorded according to others' memory, whose clinical data was with recall bias. Two of the migraine sufferers (III-7 and IV-4) were interviewed over the phone by the headache specialists. The rest of the migraine patients accepted face-to-face interviews and physical examinations. All provided informed consent, and the study was approved by the Ethics Committee of Chinese PLA General Hospital. The proband (IV-6), III-2 and III-11 have been keeping in touch with us.
Pedigree of the family and Sanger sequencing results. (a) Pedigree of the family with autosomal dominant condition. Black shading indicates the affected HM patients. The black arrow shows the proband. Diagonal line: Deceased patient. M: mutation. Genotype was marked under the family members who were under gene sequencing tests. (b) exhibits the Sanger sequencing electropherograms of the proband (IV-6), his father (III-11), his mother (III-12), his cousin (IV-2) and his aunt (III-10). The base at chr1:160105392 was mutated from G to A in IV-6, III-11 and IV-2, who are heterozygous at this location and affected by HM. III-10 and III-12 are healthy and did not carry the novel mutation. (c) Conservation of the local amino acid sequence by multiple alignment of ATPases from different species; G762S is a highly conservative position.
DNA extraction
Blood samples were taken from five migraine sufferers (II-4, III-3, III-11, IV-2, and IV-6), two unaffected consanguineous individuals (III-2, III-10) and one spouse (III-12). Genomic DNA was extracted using QIAmp 96 DNA Blood Kits (Qiagen, Hilden, Germany). The concentration of DNA was measured using Nanodrop2000 (Thermo Fisher Scientific, Wilmington, Delaware, USA).
Gene sequencing and analysis
The total DNA samples from IV-2, IV-6 and III-10 were subjected to whole-exome sequencing. Each sample generated 10.7 G data and covered 94.7% of the targeted sequence at 20 × or greater depth. First, comparative analysis was conducted on genetic data from IV-2 and IV-6 to identify variants they shared, which could narrow down the number of possible variants for further analysis by selecting the most distantly consanguineous affected members. Next, the data from III-10 was used to exclude benign variants. The remaining variants were screened by Sanger sequencing on DNA samples from II-4, III-3, III-11 and III-12. The target variant was confirmed in samples of II-4, III-3 and III-11, but was negative in III-12's sequencing.
Mutation and protein structure prediction
Algorithms to predict the impact of the novel mutation on Na+/K+-ATPase function
Prediction of G762S pathogenicity.
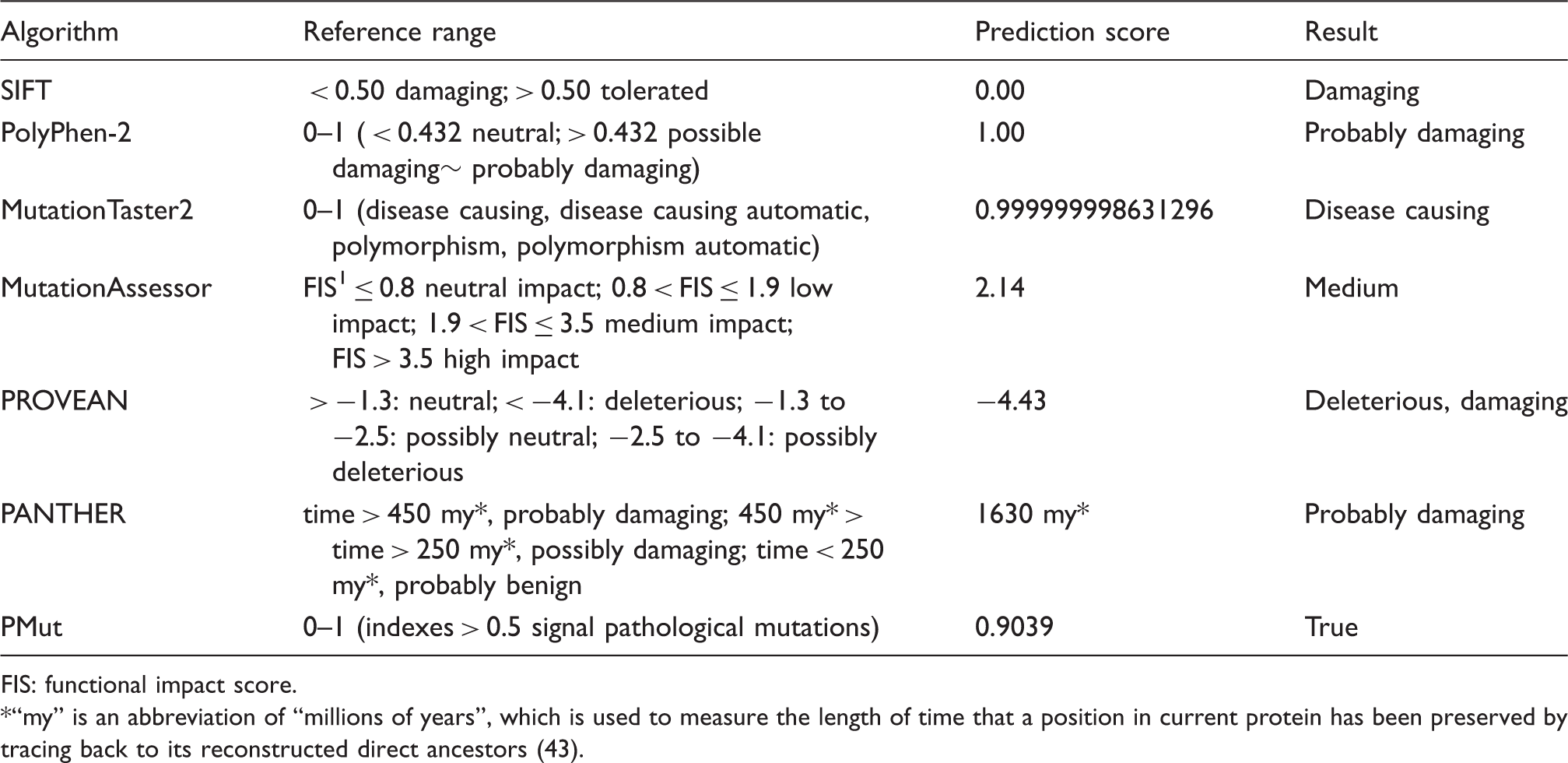
FIS: functional impact score.
*“my” is an abbreviation of “millions of years”, which is used to measure the length of time that a position in current protein has been preserved by tracing back to its reconstructed direct ancestors (43).
Structure prediction of α2 subunit of Na+/K+-ATPase due to the novel mutation
PredictProtein (24,25) and Jpred 4 (26,27) were used to compute the prediction of secondary structure changes in the Na+/K+-ATPase α2 subunit. All steps were conducted following the instructions of each software. The molecular visualization system PyMOL produced 3D images to present the changes in chemical bonds following the amino acid substitution. The reference structure 4xe5 from the Protein Data Bank (PDB), whose sequence shares 86.95% similarity with the Na+/K+-ATPase α2 subunit of Homo sapiens, was chosen for the 3D homology modeling, and the structure was confirmed at 3.9Å resolution (18). The “wizard → mutagenesis” function was used to substitute the glycine 762 with serine, and probable hydrogen bonds were deduced and added by the “polar contacts → to other atoms in object” function in PyMOL.
Functional test
Reconstruction plasmids of ATP1A2
Primers in the functional tests for ATP1A2 missense mutation G762S.

Transient expression
HEK293T and HeLa cell lines were used to test the function of mutant ATP1A2. Cells were grown in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% FBS, 2 mM glutamine, 50 units/ml penicillin, and 50 μg/ml streptomycin in a humidified, 5% CO2 atmosphere at 37 ℃. We transfected pIRES2-eGFP-ATP1A2 to the HEK293T cell line and HeLa cells separately by Lipofectamine 3000 (Cat. No. L3000015, Invitrogen). For the MTT assay, cells were seeded in 96-well dishes. Each dish was designated for testing at a set time point of 12 h, 24 h, 36 h, or 48 h.
Cell viability assay
The Methylthiazolyldiphenyl-tetrazolium bromide (MTT) assay was performed according to the instructions accompanying the assay kit (Cat. No. BB-4201-2, BestBio, China) to assess the cell viability (19). When 70% confluence was reached, the medium was changed to fresh medium containing 1 μM ouabain. At the designated time point, 10 μl of MTT (5 mg/ml) was added to each well, and the cells were incubated for another 4 h at 37 ℃. The MTT and medium were extracted, 150 μl of solvent solution was added, and the mixture was oscillated for 10 min. The optical density (OD value, proportional to the number of live cells) was evaluated using a Multiskan FC Microplate Photometer (Thermo Scientific) at 492 nm. Each transfection was performed 9–10 times at each time point.
Statistical analyses
The ratio of cell survival was calculated as followed: ratio = (OD of transfected groups/OD of non-transfected groups) × 100%. The results were given to means ± SEM, a two-tailed paired t-test was conducted (group transfected with G762S vs. group with wild-type plasmids in the same cell line) and statistical significance was set at p < 0.05. All the statistical process was performed using SPSS Version 20.0 (IBM Corp., Armonk, NY, USA). The OriginPro 2017 (OriginLab, Northampton, MA, USA) was used to produce statistical graphs.
Results
Clinical features of the family
Proband IV-6
The proband was a 22-year-old man who had suffered from HM since he was 8 years old. Attacks usually started with blurred vision, which lasted approximately 20 minutes. After that, he experienced unilateral limb numbness and weakness, which gradually spread to the same side of the face and tongue, subsequently followed by aphasia. These aura symptoms usually persisted for over one day, which dramatically impacted his life quality. A unilateral pulsating headache with nausea and vomiting developed soon after the visual aura. The headache attacks always persisted for over 24 hours and were relieved after sleep. Physical examination showed normal neurological function during the interval of attacks. Cerebrospinal fluid (CSF) analysis during the episode indicated negative results. The MRI revealed normal brain structure except one remote lesion in the right frontal lobe due to a head trauma at age 4. Electroencephalography (EEG) also appeared normal during the headache attacks. The HM attacks occurred four to six times per year. In addition, the patient also had migraine attacks without aura. He did not accept use of any prophylactic medicine due to the low frequency of attacks.
The HM pedigree
We interviewed all the living affected family members (II-4, III-3, III-7, III-11, IV-2, IV-4 and IV-6) and three unaffected members (III-2, III-10, and III-12), then depicted the pedigree (Figure 2(a)). In this four-generation family, each generation had at least one HM patient, with 10 out of 16 blood relatives affected. The sex ratio (F:M) of patients was 1:1. All the affected members suffered from HM attacks, and the clinical manifestations were highly similar. The attacks were always triggered by physical stress (cold, menstruation, fatigue) or emotional stress (heavy workload, white-coat interview, examinations at school). When the hemiplegic aura appeared, the visual disturbances, sensory abnormalities, and aphasia also presented during the aura and headache, though the appearance order of the typical auras was inconsistent. Aside from the typical auras, other brainstem auras also presented in this family: Vertigo, gait unsteadiness, tinnitus, dysarthria, hypacusis and diplopia. The duration of the aura was 0.5–2 days. The main characteristic of the headache was throbbing pain, along with nausea, vomiting, photophobia and phonophobia. The frequency of HM episodes was low in this family, six times per year at the most and once every several years at the least. Therefore, prophylactic treatment was not accepted, and they chose triptans to ease symptoms during acute attacks. The family also had other types of migraine attacks; four had attacks with common aura, and two had attacks without aura. Notably, the age of onset was decreasing with each generation (Table 3).
Mutation detection
III-7 and IV-4 were reluctant to provide bio-samples for any further tests; thus, II-4, III-3, III-11, IV-2, IV-6, and three unaffected members (III-2, III-10 and III-12) underwent genetic sequencing tests. After comparing the results of whole exome sequencing from IV-2, IV-6, and III-10 samples, a novel mutation c.2284G>A was detected in IV-2 and IV-6 and was found negative in III-10 (Figure 2(b)). The mutation was located in exon 18 of ATP1A2. The amino acid was also changed following the nucleotide mutation; the glycine at position 762 was substituted by serine (Figure 2(c)). The same mutation was also confirmed by Sanger sequencing in II-4, III-3, and III-11, who were HM patients, while it was not found in III-2 and III-12 (Figure 2(a), (b)). We screened the mutation G762S of ATP1A2 in the Database of Single Nucleotide Polymorphisms (dbSNP), Exome Aggregation Consortium (ExAC) and Human Gene Mutation Database (HGMD) to confirm that G762S was not a common base substitution and has not been reported before. In short, the clinical phenotype of the family is highly correlated to the novel mutation, and the penetrance of the pedigree is 100%. Then, we conducted software analyses and a test to evaluate the pathogenicity of G762S.
Mutation and protein structure prediction
Seven algorithms predicted the pathogenicity of G762S
We typed “ATP1A2” into the search frame of the UniProt database and picked sequences from the former 19 species on the list to do a multiple alignment of these sequences. The result showed that the glycine at position 762 was highly conserved in 19 of the species (Figure 2(c)). All seven algorithms: SIFT, PolyPhen-2, MutationTaster2, MutationAssessor, PROVEAN, PANTHER and PMut, reported that G762S was pathogenic and responsible for the familial hereditary disorder (Table 1). G762 obtained scores over the pathogenic threshold in all seven prediction systems.
G762S caused deformity in the protein structure
The PredictProtein software calculated that serine at locus 762 (S762 for short) failed to sustain the helical structure that glycine maintained (Figure 3(a)). The Jpred 4 computed that both glycine at 762 (G762 for short) and S762 could support the helical structure, but serine seemed to maintain a steadier helix condition (Figure 3(b)). PyMOL supplied a helical structure for both amino acids. The No. 01 hydrogen atom on the N-terminal of G762 formed a single hydrogen bond with the oxygen atom of G758, which was 2.0 Å long (Figure 3(c)(i)). However, S762 formed three hydrogen bonds with two amino acids, G758 and V362 (Figure 3(c)(ii)). The hydrogen atom from N-H of S762 formed a hydrogen bond with the oxygen atom from C=O of G758, while the latter formed another hydrogen bond with the hydrogen from -OH, which connected to CH2, the R group of S762. Both hydrogen bonds between S762 and G758 were 2.9 Å long. The third hydrogen bond was formed by S762 and V362, whose hydrogen was from CH2 in the R group of S762 and whose oxygen atom was from C=O of V362. The third hydrogen bond was 2.2 Å long (Figure 3(c)(ii)). The analyses from PredictProtein, Jpred 4 and PyMOL showed that G762S may cause local deformity in the secondary structure of the α2 subunit of NKA.
Secondary structure prediction of the ATP1A2 protein. (a) PredictProtein prediction results. (b) Jpred 4 prediction results. Red frame and arrows show the probable changes of G762S, compared with G762. (c) Possible 3D structure of the ATP1A2 protein. (i) The wild-type ATP1A2 protein and G762 only form one hydrogen bond with G758; (ii) the ATP1A2 protein with G762S mutation, which forms two hydrogen bonds with G758 and one hydrogen bond with V362.
G762S caused loss of function of NKA
One μM ouabain, added to the cell medium, blocked the activity of endogenous NKA, which made the MTT results show the function of transfected NKA. The transfected wild-type NKA in both HEK-293T and HeLa cell lines compensated for the loss of endogenous NKA function and enabled the cells to grow well. The transfected cells carrying the G762S mutant lost their vitality gradually, even in the first 12 hours after transfection (p < 0.01, Figure 4). It was hard to observe cell survival in transfected cells with G762S plasmids 48 hours later, while the wild-type plasmids kept transfected cells alive (p < 0.001, Figure 4).
MTT assay. (a) Condition of cell treated with ouabain 12 h after HEK293T and HeLa were transfected by pIRES2-eGFP-ATP1A2. Both G762S and WT recombinant plasmids are carrying ouabain-resistant sites. (b) The statistical results of the MTT assay indicate the cell viability after ouabain treatment.
Discussion
Over 80 mutations of ATP1A2 have been identified according to the HGMD. Most of these mutations were first reported in Europe and America (3), one was reported by South Korea (4), and one was found in Saudi Arabia (5). The prevalence of FHM and SHM is low and estimated at 0.005% (20). This Chinese FHM family is the first pedigree found with ATP1A2 mutation.
Clinical features of HM attacks in affected subjects of the pedigree.

A: aphasia; D: deceased; F: female; H: hemiplegia or hemiparesis; M: male; MA: migraine with typical aura; MBA: migraine with brainstem aura; MO: migraine without aura; S: sensory disturbance; U: unknown; V: Visual disturbance.
Comparison of several FHM2 families.

CSD: cortical spreading depression; KI mice: knock-in mice; P: Phosphorylation; TM: transmembrane structure; WB: Western blot.
The novel mutation G762S showed 100% penetrance in this family (Table 4). We detected the genotypes of five affected members, who were all G762S heterozygous. The novel mutation was not detected in unaffected members (III-2, III-10). The spouse of III-11, III-12, did not possess the G762S mutation either (Figure 2). Although FHM is a chromosome dominant disorder, the penetrance of ATP1A2 is not complete in many other pedigrees with other mutation loci. We checked five previously reported missense mutations and calculated the penetrance of the corresponding families (Table 4). The lowest penetrance was 2/3 in a German family with R763H (26). The highest penetrance was 8/9 in a family of Italian origin with G301R (25). The penetrance of a genotype is determined by genetic, environmental, and lifestyle factors, many of which are unknown and out of control of the genome (27). Members of the family with the G762S mutation resided around Shanghai and Jiangsu Province in China, where are confined downstream of the Yangtze River. The climate, habits, and customs have been relatively unchanged from generation to generation, which may be the reason for the complete penetrance in this family. The co-segregation of the mutation with the migraine phenotype in four generations and 10 affected members was complete, which implies the disease-causing potential of G762S.
We conducted seven algorithms to predict the pathogenicity of G762S (Table 1). SIFT, PANTHER, and MutationAssessor predict mutation function based on evolutionary conservation (10,12,28); PolyPhen-2 is based on alignment of sequences and the protein structure comparison strategy (29); PROVEAN is based on sequence alignment (13); PMut relies on the use of a neural network trained with a large database of neutral mutations (15); MutationTaster2 evaluates DNA sequences and examines evolutionary conservation, splice-split changes, loss of protein features and mRNA level changes (30). Different algorithms use different strategies and databases for data mining (31,32). Thus, we used seven online prediction tools to evaluate the novel mutation. As the Uniprot database showed that G762 was a highly conserved point on ATP1A2 (Figure 2(c)), all seven tools confirmed that G762S was pathogenic and G762, as a stationary residue, cannot be replaced.
The PolyPhen-2 and MutationTaster2 softwares predicted the pathogenicity of G762S and implied that the structure of the α2 subunit was changed by the amino acid substitution. We first compared the basic structure of these two amino acids: Glycine is an aliphatic amino acid with a nonpolar chain in the R group, and a hydrogen atom, while serine is a hydroxylic amino acid containing a primary alcohol group, which is polar and very soluble in water (33). It seemed inevitable that the structure of the protein would be changed by the serine mutation. Then, we compared the secondary structure of the wild-type α2 subunit and the mutant one using Jpred4 and PredictProtein. Both of the tools computed that serine changed the helical structure at site 762, but Jpred4 calculated that serine seemed to maintain a more reliable helix condition, while PredictionProtein found that serine could not sustain the helical structure at all. The amino acid residues from 760–769 are at the interface of the P domain and the TM5 (34) (Figure 1(a)), the latter of which is responsible for cation binding and transport (35). In the Uniprot database, these 10 residues are marked at the “cytoplasmic” side, which is in the loop between TM4 and TM5, and some papers have also indicated the same topology (22,36). However, some studies found that these residues reside in TM5 (7,34,37). In particular, the review written by Morth et al. indicated that R763 and L764 were at “the interface of the P domain and the transmembrane region”, but the review marked both loci in TM5 in its Table 3 (34). According to the explanation in the Uniprot Database, because the real structure of the α2 subunit of NKA has not been available, the predictions provided by the program TMHMM are not the real structure; even when “there is proof for the existence of these domains, it is difficult to determine their boundaries”. It is true that both R763 and L764 are located at the boundary of the P domain and TM5, and so is G762 (Table 4). The boundary between the P domain and TM5 is vital to the function of NKA; because it links the catalytic site of ATP and the ion transport channel to perform the physiological function of NKA. The high conservation of the sequence also implies the importance of these residues.
Hydrogen bonds are essential interatomic bonds to protein molecules, because they maintain the stiffness of proteins (38). To further analyze the deformity caused by the mutation, we used PyMOL to predict changes in the hydrogen bonds when serine substituted glycine (Figure 3(c)). G762 had only one hydrogen bond with G758 (Figure 3(c)(i)), while S762 bound G758 and V362 with three hydrogen bonds (Figure 3(c)(ii)). Generally speaking, the more hydrogen bonds, the more stable the protein (39). However, more bonds make the local structure more rigid and impact the normal folding of the protein, which can decrease the catalyzing velocity and influence the binding of cations in TM5. In short, the G762S mutation impacted the function of the α2 subunit of NKA by changing the 3D structure of the protein.
Like other mutations reported before (Table 4), G762S reduced cell survival dramatically in both HeLa cells and HEK293T cells (Figure 4), which indicated that the novel mutation was probably loss-of-function. The MTT functional test confirmed dysfunction of the α2 subunit caused by G762S, but it could not further demonstrate the changes in the catalyzing dynamics and the binding condition, which is one limitation of our preliminary research on this mutation. Different mutations on different domains would change one or several features of the protein, including the structural or functional features (Table 4). Take W887R, for example: W887 is located at the TM7-8 loop outside the cell membrane and is close to the site binding to the β subunit (Figure 1). A previous study found that R887 decreased the pump current due to the decreased level of the α2 subunit on the cell membrane, but the phenotype of knock-in (KI) mice was not apparent (8). On the other hand, G301 resides in the center of TM3, where it is close to the cation-binding pocket (Figure 1) (40). In vitro studies revealed that G301R reduced cell survival, as well as decreasing the pump current to zero (19,40); however, heterozygous KI mice showed normal protein level but susceptibility to CSD and anxious, depressed behaviors in stress condition (41,42). We also summarized the functional results of R763H and L764P, both of which are adjacent to G762S, to further deduce the possible functional changes of the novel mutation. However, both of them showed similar in vitro functional results to G301R and W887R in vitro studies (Table 4). Hence, it appears that mutations at different loci may induce similar dysfunctions of the protein in vitro. Further functional studies should be conducted on KI animal models to link the structural abnormalities to the clinical phenotypes and protein function.
In conclusion, the novel mutation we found in this family was at site 762, where the glycine was replaced by serine (G762S). Co-segregation of the mutation with the migraine phenotype in 10 affected individuals was complete, and the penetrance was 100%. Both the mutation prediction algorithms and the protein structure prediction softwares indicated that G762S was deleterious by changing the 3D structure of the α2 subunit of NKA. Meanwhile, the cell viability assay verified that G762S was a loss-of-function mutation.
Footnotes
Article highlights
This is the first FHM2 family reported in China. A novel mutation G762S in ATP1A2 was found.
Co-segregation of the mutation with the migraine phenotype in four generations of 10 affected individuals was complete, and the penetrance was 100%.
G762S was loss-of-function by changing the local structure of the interface between the P domain and TM5 of the NKA α2 subunit.
Acknowledgements
The authors would like to thank to Jiaofeng Chen and principal investigator Wei Xiong in the School of Life Sciences of Tsinghua University, who gave us guidance and great help with the experiments. Authors’ roles: ZD designed the whole project. WjT and McZ performed the mutation prediction analyses, protein structure analyses, and the manuscript writing. McZ, HxL, and ZD finished the clinical data collection and patients’ interviews. WjT and SsK conducted the functional study. EcQ amended the manuscript. SyY gave advice and supervised the conduct of the whole project.
Declaration of conflicting interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by the National Science Foundation of China (grant numbers as follows: 81471147, 81471146, 81500943, 81671077 and 81771200), Beijing Science & Technology Nova Program (grant number Z171100001117108).
*
These authors contributed equally to this work.