
Abstract
Almost all mutations in the SCN1A gene, encoding the α1 subunit of neuronal voltage-gated Nav1.1 sodium channels, are associated with severe childhood epilepsy. Recently, two mutations were identified in patients with pure familial hemiplegic migraine (FHM). Here, we identified a novel SCN1A L263V mutation in a Portuguese family with partly co-segregating hemiplegic migraine and epilepsy. The L263V mutation segregated in five FHM patients, three of whom also had epileptic attacks, occurring independently from their hemiplegic migraine attacks. L263V is the first SCN1A mutation associated with FHM and co-occurring epilepsy in multiple mutation carriers, and is the clearest molecular link between migraine and epilepsy thus far. The results extend the clinical spectrum associated with SCN1A mutations and further strengthen the molecular evidence that FHM and epilepsy share, at least in part, similar molecular pathways.
Introduction
Both migraine and epilepsy are complex episodic neurological diseases with genetic and environmental factors playing a role in their pathogenesis (1, 2). Epidemiological studies have indicated that there is a clear bidirectional increased risk of migraine and epilepsy (3). This suggests that migraine and epilepsy, at least in part, may share pathophysiological mechanisms (4).
Most genes that are involved in rare monogenetic forms of epilepsy encode subunits of channels transporting sodium, potassium or chloride ions (for a recent review see (2)). Gene identification in migraine has been successful only for familial hemiplegic migraine (FHM), a rare, monogenic, autosomal dominant subtype of migraine with aura with unilateral motor weakness during the aura phase (5). All three FHM genes identified thus far are involved in ion transport (6). The FHM genes encode subunits of CaV2.1 calcium channels (CACNA1A: FHM1) (7), sodium-potassium ATPases (ATP1A2: FHM2) (8) and NaV1.1 sodium channels (SCN1A: FHM3) (9).
Despite their comorbidity, co-occurring migraine in epileptic mutation carriers has not been mentioned, probably because migraine in a patient or family with epilepsy will often be regarded coincidental because of the high prevalence of migraine in the general population. Epilepsy, however, has specifically been reported in some FHM mutation carriers (10). In FHM1 mutation carriers, seizures may occur during severe hemiplegic migraine attacks (11). Seizures have been described in two FHM1 I1710T mutation carriers in one family independently of hemiplegic attacks (12) and causing status epilepticus during hemiplegic migraine attacks in a sporadic patient (13). Several carriers of FHM1 mutation S218L, which is associated with severe FHM attacks triggered by trivial head trauma, had epileptic seizures (14, 15). In FHM2 mutation carriers various types of childhood and adult epilepsy have been reported with different FHM2 mutations (8, 16–18). In rare cases, epilepsy is part of particularly severe phenotypes with alternating hemiplegia, coma with permanent cerebellar signs or mental retardation (19–21).
A recent molecular link between migraine and epilepsy has come from the identification of the first two FHM mutations in SCN1A, a well-known epilepsy gene (9, 22). Over 150 mutations in the SCN1A gene have been reported with generalized epilepsy with febrile seizure plus additional symptoms (GEFS+) and severe myoclonic epilepsy of infancy (SMEI) (23, 24). Both diseases are severe epileptic phenotypes occurring in childhood. The majority of SMEI mutations occur de novo and are either nonsense or frameshift mutations resulting in protein truncation and consequent loss-of-function or missense mutations with a wide range of functional consequences (25). The milder GEFS+ phenotype is mostly caused by missense mutations, showing either loss- or gain-of-function effects (26). Up to now, no associated migraine has been reported for any of the epilepsy mutations. It is not known why most SCN1A mutations cause childhood epilepsy, whereas only two cause hemiplegic migraine. A clinical continuum of migraine and epilepsy is probably associated with SCN1A mutations as a result of altered neuronal excitability (27).
Here we describe the first SCN1A mutation with several mutation carriers with partly co-segregating FHM and epilepsy, which provides additional support for the clinical continuum hypothesis. Our results underscore the complex relation of migraine and epilepsy and provide further molecular evidence that the two diseases share common pathways.
Methods
Clinical description
We investigated a Portuguese FHM family with partially co-segregating epilepsy (Fig. 1A). All subjects were interviewed and the clinical headache diagnoses were established according to International Headache Society criteria (International Classification of Headache Diseases criteria) (5), whereas seizures were classified according to the criteria of the International League Against Epilepsy (28). Diagnoses were made prior to genetic analyses by a neurologist experienced in migraine and epilepsy. This study was approved by the ethics committee of the Hospital Geral de Santo António, Porto. All individuals gave written informed consent.
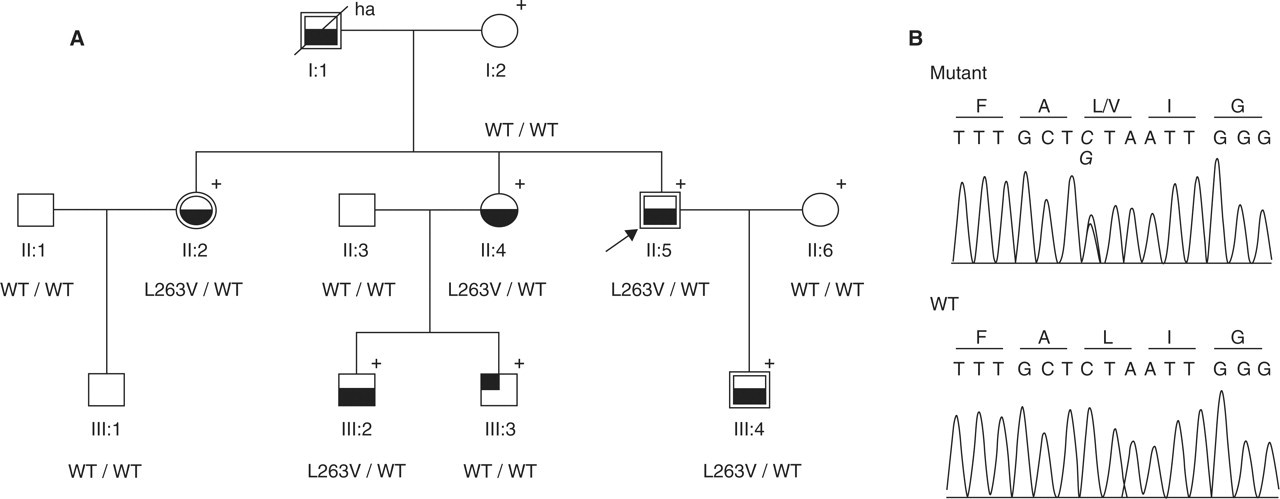
Pedigree of the Portuguese family with familial hemiplegic migraine (FHM) and epilepsy. (A) Symbols with lower half filled represent individuals with FHM; the individual with the upper-left quarter filled has migraine without aura; double lined symbols represent individuals with epileptic seizures. The arrow indicates the proband. The ‘+’ shows individuals that were clinically evaluated; ‘ha’ indicates that clinical information was obtained heteroanamnestically. The presence of the heterozygous mutation is indicated by L263V/WT and the absence by WT/WT (wild type). (B) Electropherogram of the relevant part of exon 6 showing the heterozygous C→G nucleotide change (L263V) in the proband (Mutant) and healthy subject (WT) is shown.
Clinical details of affected family members are shown in Table 1. Six patients suffered from typical hemiplegic migraine attacks, with an age of onset varying from 10 to 18 years and an attack frequency of less than one to three per year. None of the patients reported (inter)ictal cerebellar abnormalities. Generalized tonic-clonic seizures (and in one case additional complex partial seizures) were co-segregating with hemiplegic migraine in three family members (i.e. II:2, II:5 and III:4), and probably in one deceased person (I:1) [seizures similar to those of the other family members were reported by his wife (I:2)]. Onset of seizures was between 4 and 8 years. Interictal EEG recording was unremarkable in patients II:2, II:5 and III:4. Structural lesions in these patients were excluded on the basis of computed tomography (i.e. II:2 and II:5) or magnetic resonance imaging (i.e. III:4) investigations (data not shown). Febrile convulsions were not reported in any of the family members. Epileptic seizures occurred independently from FHM attacks, and the age at onset was generally somewhat later than for the FHM attacks. Treatment with a low daily dose (400 mg/day) of carbamazepine in patients II:2 and II:5, who had a body weight of < 60 kg, was successful with respect to both epileptic and hemiplegic attacks as patients remained attack free. Patient III:4 occasionally had epileptic attacks when treated with a daily dose of 600 mg of carbamazepine, but he did not tolerate a higher dose. Psychomotor development of all individuals was normal.
Summary of the clinical features of SCN1A mutation carriers

∗Characteristics of aura: visual symptoms/sensory symptoms/hemiplegia/aphasia; −, absent; +, present; GTC, generalized tonic-clonic seizure; CP, complex partial seizure; NA, not applicable.
Haplotype analysis
Genomic DNA was isolated from peripheral leucocytes using a standard salting-out extraction method (29). The involvement of the FHM1 (19p13) locus was excluded on the basis of genetic markers D19S221, D19S1150, D19S226 as described before (7). In addition, involvement of the FHM2 and FHM3 loci was investigated by analysing genetic markers D1S2624, D1S2707, D1S2844 (FHM2; 1q23) (16) and D2S156, D2S2330 and D2S2381 (FHM3; 2q24) (9), respectively (data not shown). Oligonucleotide primer sequences were obtained from the Human Genome Database (http://www.gdb.org/). Polymerase chain reaction (PCR) amplification was performed using standard conditions. PCR products were detected on an automated fragment run analyser (ABI 3700 DNA sequencer; Applied Biosystems, Foster City, CA, USA). All genotypes were analysed and independently scored by M-J.C. and K.R.J.V. using Genescan and Genotyper 2.1 software (Applied Biosystems). Haplotype was constructed evaluating the segregation and assuming a minimal number of recombinations.
Mutation screening
All exons and flanking intronic regions of the ATP1A2 and SCN1A genes were amplified by PCR, using genomic DNA of the proband as a template. Direct sequencing was performed on PCR products using Cycle Sequencing (Prism Big Dye Terminators Cycle Sequencing kit; Applied Biosystems) and an ABI3700 DNA sequencer (Applied Biosystems). Sequencing was performed in the forward and reverse direction. For detection of the SCN1A mutation L263V (C→G, nt position 787, Ac. no. NM_006920), exon 6 was amplified by PCR using primers ‘E6F’ (5′-TTGCTTCTCCACTAGCGTTG-3′) and ‘E6R’ (5′-ACTTGAGGGGCTGGATATCC-3′), resulting in a 489-bps product that was subjected to direct sequencing. One hundred and fifty healthy controls (i.e. Portuguese blood donors without migraine history) were screened for the SCN1A mutation by direct sequencing.
Results
Haplotype analysis in the Portuguese family was compatible with both the FHM2 (i.e. 1q24) and FHM3 (i.e. 2q24) locus as single haplotypes co-segregated with disease in all five FHM patients for which DNA was available (data not shown). Sequence analysis of the ATP1A2 gene did not reveal a causative mutation. In contrast, in the SCN1A gene a novel heterozygous missense mutation was identified, changing a single nucleotide in exon 6 (nt 787 C→G) (Fig. 1b). This C to G nucleotide transversion replaces a leucine for a valine at position 263 of the NaV1.1 α1 subunit (Fig. 2A). The L263V mutation co-segregated with hemiplegic migraine in all five FHM individuals and was absent in 300 control chromosomes. Notably, three of the five FHM patients with the L263V mutation also had epileptic seizures. Patient I:1 most probably is the fourth patient from this family suffering from hemiplegic migraine and epilepsy, but no DNA was available to confirm the presence of the L263V mutation. No other associated neurological symptoms such as ataxia were reported for any of the family members. Taxonomy analysis showed a strong evolutionary conservation of Leu263 amino acid among different voltage-gated sodium channels α subunits (Fig. 2B).

Location of FHM3 SCN1A mutations and conservation of Leu263. (A) Schematic representation in the Nav1.1 α1 subunit with the location of the FHM3 mutations. (B) Evolutionary conservation of the relevant part of the D1S5 transmembrane segment. Amino acid numbering for each sequence is presented. The grey box (with L) represents conservation of residue Leu263. Protein accession numbers are as follows: Homo sapiens: SCN1A (P35498), SCN2A2 (Q99250), SCN3A (Q9NY46), SCN4A (P35499), SCN5A (Q14524), SCN7A (Q01118), SCN8A (Q9UQD0), SCN9A (Q15858), SCN10A (Q9Y5Y9), SCN11A (Q9UI33); Rattus norvegicus: SCN1A (P04774); Gallus gallus: SCN1A (ENSGALP00000017793) and SCN3A (ENSGALP00000017960); and Takifugu rubripes: SCN8A (NEWSINFRUP00000180009). All sequences were retrieved from NCBI or Ensembl databases.
Discussion
Here we report a novel L263V missense mutation in the SCN1A gene encoding the α1 subunit of voltage-gated sodium NaV1.1 channels in a Portuguese family with FHM and partially co-segregating epilepsy. Several factors indicate that L263V is the causative mutation in this family with FHM and epilepsy: (i) L263V co-segregated in all five patients with hemiplegic migraine, three of them also had epilepsy (a fourth, deceased person probably had the mutation and epilepsy); no phenocopies or cases of incomplete penetrance were observed; (ii) L263V was absent in 300 control chromosomes; and (iii) Leu263 shows strong evolutionary conservation among several voltage-gated sodium channel subunit homologues across species; and (iv) its location in transmembrane domain DIS5 of the protein suggests an important functional role.
α1 subunits of NaV1.1 sodium channels consist of four homologous domains (DI–DIV), each containing six highly conserved transmembrane segments (S1–S6), which form the central pore of the sodium channel (30). Mutation L263V is located in the highly conserved S5 segment of the first domain (DIS5). S5 and S6 segments and S5-S6 linkers line are important for ion selectivity and gating kinetics of the channel (31–32). No particular functional role has been established for DIS5, but closely located mutation I252N has been found associated with SMEI (33). Various other missense SCN1A mutations have been identified in S5 segments and are associated with different epileptic phenotypes of GEFS+ (34, 35), SMEI (36, 37) or SMEB (37–39).
The L263V mutation is the third FHM3 mutation. The first two FHM3 mutations, Q1489K and L1649Q, were identified in large German and US families, respectively, and are associated with FHM without additional neurological features such as epilepsy or ataxia (9, 22). Only in three of the 18 Q1489K mutation carriers with hemiplegic migraine were seizures reported that occurred only during infancy [two patients (in different families) with one episode; one patient with more attacks] (9). The Portuguese family with the L263V mutation displays co-occurring hemiplegic migraine and generalized tonic-clonic epilepsy with a high penetrance. It is not clear how this mutation can cause epilepsy in certain attacks and hemiplegic migraine in other attacks. A detailed analysis of the functional consequences of the L263V mutation and the two other FHM3 mutations is underway to answer that question.
In conclusion, this study has strengthened the molecular link between migraine and epilepsy, supporting the existence of a continuum of chronic episodic disorders. In addition, it has expanded the clinical spectrum associated with FHM3 SCN1A mutations.
Footnotes
Acknowledgements
We thank our Portuguese FHM family for their dedicated participation in this study. The main financial support for this work was provided by the European Community, through a Marie-Curie grant for PhD students (M-J.C.; contract no. QLGA-CT-2000-60005). Additional support was obtained from grants of the Netherlands Organization for Scientific Research (NWO) (903-52-291, M.D.F., R.R.F.; and Vici 918.56.602, M.D.F.), The Migraine Trust (R.R.F., M.D.F.), the EU ‘EUROHEAD’ grant (LSHM-CT-2004-504837; M.D.F., R.R.F., A.M.J.M.v.d.M.). This study was supported by the Centre for Medical Systems Biology (CMSB) in the framework of the Netherlands Genomics Initiative (NGI).