
Abstract
Objective
To review and discuss the literature on the role of cortical structure and function in migraine.
Discussion
Structural and functional findings suggest that changes in cortical morphology and function contribute to migraine susceptibility by modulating dynamic interactions across cortical and subcortical networks. The involvement of the cortex in migraine is well established for the aura phase with the underlying phenomenon of cortical spreading depolarization, while increasing evidence suggests an important role for the cortex in perception of head pain and associated sensations. As part of trigeminovascular pain and sensory processing networks, cortical dysfunction is likely to also affect initiation of attacks.
Conclusion
Morphological and functional changes identified across cortical regions are likely to contribute to initiation, cyclic recurrence and chronification of migraine. Future studies are needed to address underlying mechanisms, including interactions between cortical and subcortical regions and effects of internal (e.g. genetics, gender) and external (e.g. sensory inputs, stress) modifying factors, as well as possible clinical and therapeutic implications.
Keywords
Introduction
Migraine is a common debilitating brain disorder characterized by recurring attacks of moderate to severe headache (1), which are often accompanied by other neurological symptoms such as an enhanced sensitivity to light, sound, touch and smell (2). There are different subtypes of migraine, with overlapping but also distinct clinical features (1). For example, one third of migraine patients experience transient neurological symptoms preceding some of their attacks, the so-called migraine aura, which is characterized by visual, tactile, motor and/or speech disturbances.
There is strong and long-standing evidence for cortical involvement in aura pathophysiology. Already in the 1940s, Leao (3) and then Milner (4) proposed that the clinical phenotype of migraine aura may be secondary to a propagating cortical phenomenon, cortical spreading depolarization (CSD), a slowly spreading cortical wave of neuronal and glial depolarization followed by suppression of activity (5,6). Initial support for this concept came from cerebral blood flow measurements in migraine patients, which showed spreading oligemia during migraine attacks with aura (7–9). These findings were confirmed and extended in 2001, when Hadjikhani et al. provided human imaging evidence of retinotopic congruence between visual aura perception and CSD-typical changes in the blood-oxygen-level dependent (BOLD) signal (10) in the occipital cortex of a migraineur during his aura.
Recent studies support migraine as a neuronal network disorder, involving integrated activities across subcortical and cortical brain circuits that are important in head pain and sensory processing (11). The headache phase involves activation of the trigeminovascular system that conveys nociceptive information from the meninges to central brain areas and the cortex (12) (Figure 1). Although the role of the cortex in initiation of headache is not completely understood, it is clear that the cortex contributes to modulation and representation of head pain as well as amplification of sensory inputs (11). Additional network circuits implicated in migraine pathophysiology include thalamo-cortical, hypothalamic, as well as those involving brainstem and trigeminal ganglia, based on evidence for altered cellular biochemical or bioelectrical properties, microstructures, and functional connectivities (11–18).
Schematic representation of the cortex in relation to the trigeminovascular pain pathway. Cortical spreading depolarization (CSD) is the likely neurophysiological phenomenon underlying the migraine aura and consists of a slowly propagating wave of cortical network depolarization. CSD-associated rise in potentially noxious molecules including K+ and low pH in the extracellular space (light yellow shaded) may reach pial, arachnoid, and dural surfaces and activate the perivascular sensory afferents from the trigeminal ganglion (TG) neurons. Signals of activated meningeal nociceptors are relayed through TG nerve processes to the trigeminal cervical complex (TCC) in the brainstem and subsequently to thalamic and cortical areas to produce the sensation of pain. The cingulate cortex (CC), situated in the medial aspect of the cerebral cortex and involved in emotional and affective processing receives input from the thalamus and several regions of the frontal, parietal, and temporal cortex.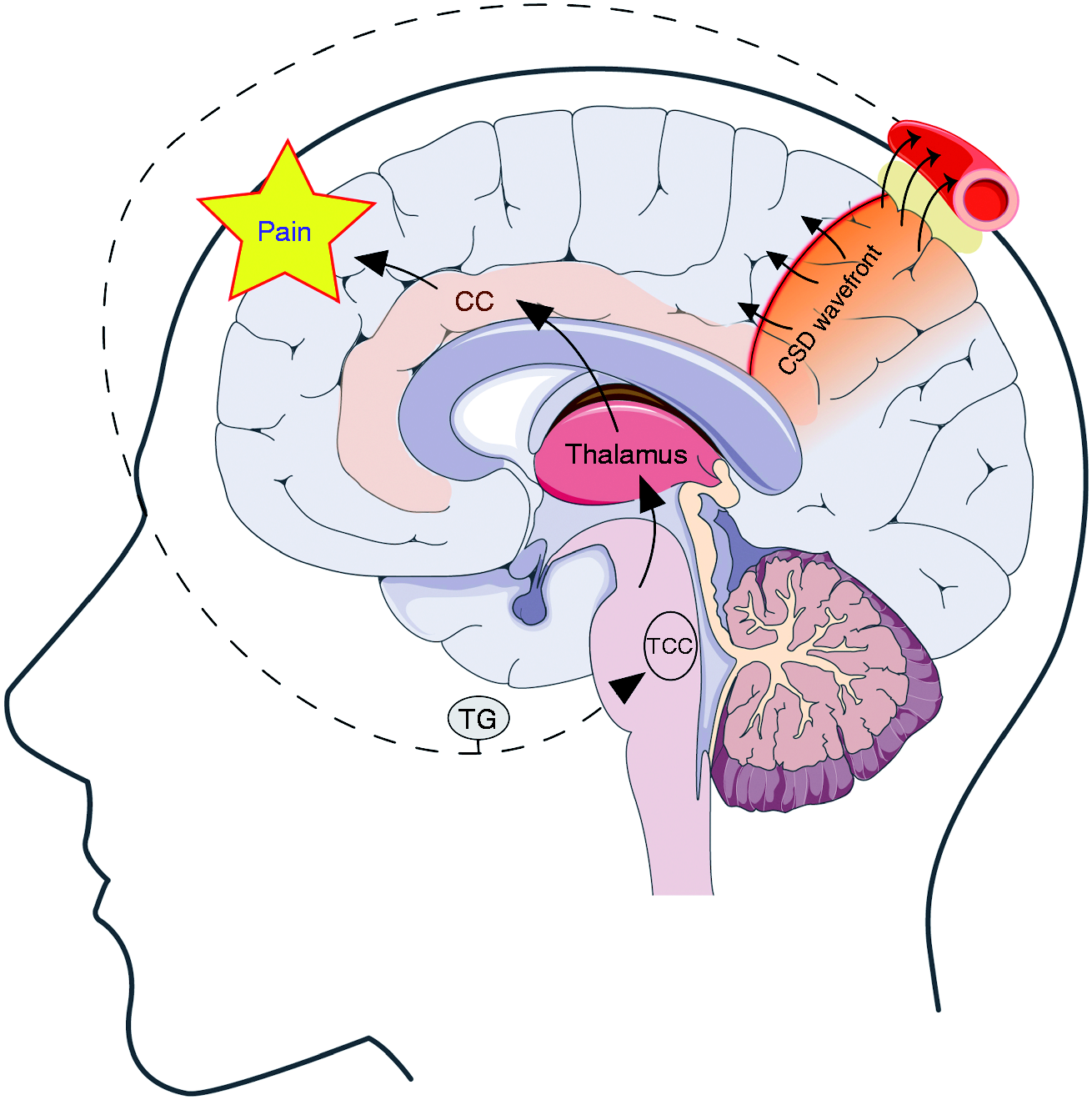
The focus of this review is to discuss structural and functional findings in the cortex of migraine patients and related animal models, with emphasis on neuronal network changes. Integration of clinical and preclinical findings on cortical alterations in migraine can help understand the pathophysiological changes underlying attack susceptibility as well as chronification of migraine, and may aid in the development of novel diagnostic and therapeutic strategies.
Clinical neuroimaging studies on cortical structure and function in migraine
Neuroimaging is a useful non-invasive tool to investigate mechanisms underlying migraine. Imaging techniques can be divided into structural imaging, which provides anatomical information and functional imaging that obtains physiological information; for example, on blood oxygenation (BOLD magnetic resonance imaging (MRI)) or blood flow (single-photon emission computer tomography (SPECT)), metabolism (positron-emission tomography (PET)) as well as alterations of neurotransmitters or their receptors (PET), MR spectroscopy (MRS). Combining the static and dynamic information of these imaging studies can help interrogating the complex pathophysiology of migraine, and novel computational models can provide tools for classification between migraine subtypes with possible implications in guiding and monitoring therapy. Figure 2 summarizes the key imaging findings discussed in the paragraphs below.
Summary of the key observations from clinical neuroimaging studies on migraine and schematic representation of cortical regions (see Figure 1 for location of the cingulate cortex). See main text for details.
Neuroimaging findings on cortical structure in migraine
Structural data from migraine patients obtained during the interictal phase indicate changes in cortical sensory and affective processing regions in comparison to controls (19). Increased cortical thickness has been reported in the somatosensory cortex (S1) in patients with episodic migraine with or without aura (20–22), especially in the area representing the face in the post-central gyrus (22), suggesting an adaptive change of this cortical region upon repeated sensory drive. Increased thickening of the left middle frontal sulcus and left temporo-occipital incisure, including visual processing areas, has also been reported, with no differences between migraine with and without aura (23). Increased cortical thickness in migraineurs may be a trait resulting from a plastic change, and/or may represent reactive gliosis or neurogenesis in response to repeated migraine attacks (24). However, other studies revealed contradicting results. For example, Datta et al. showed no changes in cortical thickness in patients with migraine with or without aura (25), while others showed decreased grey matter volume in the superior temporal gyrus, inferior frontal gyrus, and precentral gyrus (26) as well as in visual areas V3 and V5 of the right occipital cortex (27). The latter study observed reduced grey matter in visual cortex to relate with increased volumes in the angular, middle temporal, precentral and superior frontal gyrus, and lateral geniculate nucleus, suggesting inherited or acquired structural changes in visual processing pathways (27). Discrepant findings with other studies might be explained by the fact that grey matter volume reflects not only cortical thickness but also additional features such as cortical folding and surface area.
With respect to the clinical phenotype, reduced grey matter volume in the anterior cingulate cortex and insula (22) was associated with increased headache frequency, suggesting that affective processing regions may display an adaptive response to repeated migraine attacks. Similar observations were made for the frontal cortex (28) and visual area V5 (27), suggesting impaired executive and visual function with repeated attacks. Consistent with these observations, a meta-analysis for voxel-based morphometric studies from 1990 to 2014 showed reduced grey matter volumes in the middle and inferior frontal as well as pre-central cortex, sub-lobar insula, sub-gyral temporal cortex, and temporal cortex in patients with migraine (29), suggested to reflect cognitive, emotional, and autonomic changes in patients with migraine. The degree of reduction in grey matter volume (cingulate cortex and multiple brain regions in the frontal, temporal and occipital lobes, precuneus and cerebellum) seemed to be more profound in patients with chronic migraine (26,30). In contrast, a recent study showed increased grey matter volume in the amygdala and putamen in patients with chronic migraine (31). Taken together, these regional differences of grey matter volume may suggest maladaptive cortical plasticity changes related to attack chronification (11,32).
Structural and regional changes in the cortex may provide diagnostic tools for classifying migraine subtypes. By incorporating morphometric imaging features into principal component analysis, including cortical surface area, cortical thickness, and regional volumes, classifiers were able to differentiate patients with chronic migraine from those with episodic migraine or healthy controls, and could accurately predict chronicity of migraine. The multivariate models involved structural measures of the temporal pole, anterior cingulate cortex, superior temporal lobe, entorhinal cortex, medial orbital frontal gyrus, and pars triangularis that were identified as having abnormal structure and/or function in patients with migraine (33).
Cortical functional imaging during the interictal migraine phase
Given the difficulty of predicting the time of attack onset, interictal imaging studies are easier to perform and far more common than studies during the other stages of migraine. Most functional imaging studies suggest hyperexcitability in cortical sensory regions during the interictal phase, consistent with dysfunctional pain processing and multisensory integration playing an important role in migraine pathogenesis (11,12).
Task-based functional MRI in patients with migraine has been studied most extensively in response to visual stimuli. For the primary visual cortex, enhanced BOLD responses to visual stimuli during the interictal phase were observed for migraine with but not without aura, despite similar interictal visual discomforts, supporting the view that visual cortical hyperresponsiveness may be related to visual aura susceptibility (34). A PET study revealed visual stimuli to activate the occipital cortex during the interictal phase in migraineurs but not in control subjects (35). Task-based studies utilizing stimuli other than visual have been investigated less, but similarly suggest increased cortical activation in migraineurs, with activation of abnormal cortical regions by painful or noxious stimuli (14). For example, increased BOLD activation of the primary somatosensory cortex has been shown in patients with migraine during a motor task (36). Similarly, an enhanced BOLD response in the temporal pole, a multisensory region that integrates visual, auditory, olfactory and somatosensory stimuli, has been shown in patients with migraine after painful heat stimuli (37). In addition, PET showed higher activation in the temporal pole upon olfactory stimulation in patients with migraine (38). Interestingly, patients with high-frequency migraine attacks, compared to patients with low frequency attacks, showed stronger somatosensory cortex activation in response to noxious stimulation (22).
An MRS study found high resting lactate levels in the visual cortex in migraineurs with aura that was even further increased upon visual stimulation in patients with complex auras (39), suggesting possible underlying mitochondrial dysfunction and stimuli-evoked abnormal metabolic strain in aura patients. Impaired energy metabolism in the occipital cortex was also suggested by MRS studies for migraine without aura patients (40,41). Studies using 1H-MRS suggest increased levels of the excitatory neurotransmitter glutamate in the visual cortex of migraineurs, with higher interictal ratios of glutamine/creatine (42), glutamine/GABA (43), glutamate/glutamine (44), and enhanced glutamate/creatine levels during visual stimulation. In contrast, the GABA/creatine ratio (45) was reduced in the occipital cortex of episodic migraineurs, whereas in chronic migraine reductions in N-acetyl-aspartate were observed in this region (46). With high magnetic (7T) 1H-MRS and diffusion-weighted spectroscopy, glutamate levels were found to be increased in both primary and secondary visual cortex in migraineurs with, but not without aura (47). This observation is in line with the concept that increased cortical glutamatergic activity contributes to increased cerebral excitability and enhanced susceptibility to CSD, as has been observed for familial hemiplegic migraine (FHM) (48).
Assessment of neuronal networks may help in deciphering mechanisms underlying migraine. Important components of the head pain matrix and somatosensory cortex showed increased resting-state functional connectivity with the periaqueductal gray matter (PAG), while connectivities between the prefrontal cortex, anterior cingulate, and amygdala with PAG decreased along with increased headache frequency (49). The anterior insula, a region important for emotional salience, showed heightened interictal intrinsic connectivity with primary sensory networks and the pons in migraine without aura (50). However, connectivity between the anterior insula and occipital areas was reduced in migraine patients with, but not without aura (51). This reduced connectivity correlated with headache severity (51), in line with reduced functional connectivity including the anterior insula and occipital regions for chronic migraine (52). In chronic female migraineurs, coherence of salience, central executive and default mode networks was reduced (53). Interestingly, stronger functional connectivity of the anterior cingulate cortex with the frontal pole and temporal pole was seen in episodic migraineurs with high frequency attacks (22). Together, these observations may suggest maladaptive cortical network plasticity changes to contribute to, and/or be a consequence of, repeated attacks.
Cortical functional imaging data from the premonitory, aura and ictal phase of migraine
Mostly by serendipity or via longitudinal study design, functional imaging studies were performed during the preictal or ictal stages of a spontaneous migraine attack (14,54). An alternate and commonly employed way to study the preictal or ictal phase of attacks is to provoke attacks by experimental migraine triggers such as nitroglycerin infusion (55), pituitary adenylate cyclase-activating polypeptide (PACAP) (56) or calcitonin gene-related peptide (CGRP) (57), although it remains disputable whether the (cortical) mechanisms underlying attack initiation are similar to those of spontaneous attacks (58).
In a patient with migraine without aura who received fMRI for 30 consecutive days, the visual cortex showed enhanced BOLD responses following a noxious nasal stimulus during preictal and postictal phases, in comparison to the interictal phase, suggesting dynamic changes in visual cortical excitability (59). Using resting-state fMRI, several cortical regions including sensory and pain processing areas showed altered connectivity prior to or during attacks (60). During the initial 6 hours of a spontaneous migraine attack, stronger functional connectivity was observed between the medial prefrontal cortex, posterior cingulate cortex and insula, with the strength of functional connectivity between medial prefrontal cortex to insula being negatively correlated with headache pain intensity (61). During attacks, connectivity was increased for connections of pain processing regions (including the parietal, insular, primary motor and orbitofrontal cortex) with contralateral parts of the thalamus (62), and between the primary somatosensory cortex and pons, as well as visual area V5 and the middle frontal gyrus ipsilaterally (63). Reduced functional connectivities were reported for cortical regions involved in executive, cognitive and attention networks, including the frontal gyrus, cingulate and temporal cortices (60).
Using fMRI and noxious heat stimuli, activation of the anterior temporal pole was more pronounced in the ictal than the interictal state (37). The possibility that structural changes can occur in cortical regions during attacks is suggested by a voxel-based-morphometry study of T1-weighted MRI scans in migraine without aura, which showed increased grey matter densities within the left temporal pole and bilateral insula during attacks (64).
During spontaneous migraine attacks, a PET study revealed increased activation in pain processing cortical areas including the anterior and posterior cingulate cortex, insula, prefrontal cortex, and temporal lobes along with other pain related structures such as the dorsal pons and thalamus (65). Stronger activation, indicative of hyperexcitability, was also found in the visual cortex upon low luminous stimulation in the ictal stage of spontaneous migraine attacks (66). For nitroglycerin-induced migraine headache, occipital lobe activation was noted only during the early and late premonitory and not the ictal phase, whereas temporal, frontal and parietal cortical hyperactivity were observed for the early premonitory phase only. During the ictal phase, activation was noted at the right precentral gyrus, right prefrontal gyrus, left post central gyrus, and right parietal lobe (67). Interestingly, patients experiencing photophobia during the premonitory phase following nitroglycerin showed stronger activation of the extrastriate visual cortex (Brodmann area 18) than patients without photophobia (68).
How precisely the cortex may adapt in relation to repeated migraine attacks remains unsettled. It is likely that differences in attack frequency (22), sex (69), peripubertal changes (70) or drug treatment (71) may also contribute to observed plasticity changes.
Imaging studies performed during the aura phase of attacks have provided important insight into the phenomenon of CSD in humans, based on CSD-related neurovascular and metabolic changes. CSD is a wave of neuronal and glial depolarization that spreads with a velocity of 3–6 mm/min across the cerebral cortex, and is the electrophysiological event underlying migraine aura. The depolarization wave is typically followed by transient suppression of neuronal activity. During CSD, all major ions show abrupt and profound changes in the intra- and extracellular compartment. Neurons swell because water influx follows the influx of Na+ and Ca2+ ions. CSD propagation is generally regarded as a reaction/diffusion process. Thus, neurons release neuroactive substances such as K+ or glutamate, which diffuse to adjacent neurons and trigger a self-propagating regenerative process (72–74). The vascular response to CSD in otherwise metabolically intact tissue includes a short-lasting hyperemia of approximately 2 minutes followed by a prolonged decrease of regional blood flow for up to 2 hours (72,75). During the latter, neurovascular coupling is impaired, and functional activation is attenuated (76), a phenotype also described in migraineurs (77). In patients with evoked migraine aura, a 133Xe SPECT study demonstrated spreading cortical oligemia suggestive of CSD (78). A BOLD fMRI study for visually-triggered migraine demonstrated that the onset of headache was preceded by suppression of initial cortical activation, which slowly propagated into the contiguous occipital cortex at a rate ranging from 3–6 mm/min (79). In a pivotal study, BOLD signal transients resembling CSD, developing within the extrastriate cortex (area V3A) and propagating along the visual cortex, were retinotopically congruent with the patient's visual percept of aura (10). Consistently, a perfusion-weighted imaging study demonstrated a significant reduction in relative cerebral blood flow in the occipital cortex contralateral to the affected visual hemifield (80,81). Intriguingly, a spreading oligemia-like phenomenon has also been found in a patient during a spontaneous migraine without aura attack using PET (8).
While a recent case report suggests that an aura can be clinically silent (82), it remains inconclusive whether SD (either cortical or subcortical) actually occurs during migraine attacks without aura and remains asymptomatic. Both for attacks with and without aura, direct evidence for CSD or subcortical SD as a trigger for headache in patients is lacking. It is possible that migraine patients display a generally reduced threshold for activation of the trigeminovascular system, which could be independent from the propensity to develop CSD (83–85). The concept that a single CSD activates headache mechanisms in migraine patients is challenged, for example, by observations from ischemic stroke. While multiple CSD events are detected in ischemic stroke patients undergoing EEG recordings, stroke-related headache has been reported only for a relatively small number of patients (83).
Clinical neurophysiology studies on cortical function in migraine
Neurophysiological studies using scalp electroencephalography (EEG) in migraine patients (86–89) have yielded contradictory results of hyper-as well as hypo-excitability. Given the dynamic network interactions underlying attack susceptibility (11,12), discordant findings in EEG studies may reflect differences in cortical and subcortical network responses to various types of stimulation (87,88,90). Using EEG, it is not possible to precisely discern contributions of excitatory versus inhibitory, or cortical versus thalamic networks without using additional pharmacology or neuromodulatory approaches. Targeted cortical stimulation by transcranial magnetic stimulation (TMS) provides a solution to non-invasively influence and thereby study cortical excitability directly (90–93). Alternative approaches to identify activity changes specific to the cortex include high spatial resolution magnetoencephalography (MEG) (94). Key neurophysiological findings discussed below are indicated in Figure 3.
Summary of the key observations from clinical neurophysiological studies on migraine. See main text for details.
Cortical neurophysiological changes during the interictal phase
Visual inspection of interictal EEG reveals predominant normal-interval EEG records in migraineurs, whereby mild interictal EEG abnormalities reported include increased power in the delta band – also with respect to the painful fronto-central region (95) – alpha, beta and theta bands, and interhemispheric alpha-band asymmetries (86,96,97). For the occipital cortex, lower interhemispheric alpha coherence (98) and altered beta band activity (99) were specifically related to patients with aura, suggesting particular plasticity changes in this region.
Visual evoked potentials (VEPs) have been widely studied as indirect readout of visual cortex responsivity (88), whereby several studies reported no change (100) or a reduction (101,102) in VEP amplitude interictally, in line with unaltered (103) or reduced auditory (104) and somatosensory responses (105,106). Enhanced cortical responsivity between attacks is also reported, using visual (88), auditory (107), heat (108,109) or affective picture stimulations (110,111). A combined PET/VEP study recently related enhanced VEP responses to decreased glucose uptake in occipital areas in migraineurs without aura (112), which may suggest activity-induced disturbance of cerebral metabolic homeostasis contributes to attack susceptibility. For migraine with compared to without aura, VEP responses were reportedly enhanced (113–116) or unchanged (117,118). An enhanced cortical response to prolonged visual stimulation (photic drive) between 10 and 20 Hz in migraineurs again suggests plasticity changes involving the primary visual cortex (116,119,120). Attenuation (97) or variation (121) of photic drive in other studies could reflect complex (sub)cortical network changes upon dynamic visual triggers (122). For repeated low-frequency stimuli, impaired normal habituation response has been suggested as another reflection of enhanced cortical responsivity (87). This “lack of habituation” has been reported interictally for visual, auditory, somatosensory, nociceptive and cognitive triggers (93,123–125), but was not consistently seen in all studies (100,113,126,127), leaving debate on this parameter as a benchmark of cortical hyperresponsivity (120,127,128).
To localize neurophysiological findings to the cortex, source localization using MEG revealed enhanced theta band connectivity in the occipital cortex in between attacks for migraine with compared to without aura (129). Using somatosensory stimulation, enhanced primary somatosensory cortex responses in migraineurs correlated with attack frequency (130), in line with an association of increased somatosensory responsivity and migraine chronification for migraine without aura (131). Using visual stimulation and MEG, chronic migraine patients displayed a persistent visual cortex excitability pattern comparable to that of the ictal state in episodic migraine patients (132).
As an alternative approach to study cortical function more directly, TMS can be used to modulate cortical function during functional readouts. Based on visual phosphene perception as indirect readout following TMS over the occipital cortex (133), despite conflicting observations in earlier studies (89), decreased thresholds were reported in a meta-analysis in migraine with and without aura that became specific to migraine with aura with more localized stimulation (92). TMS studies on motor cortex excitability, using muscle responses as indirect readout, noted decreased (134,135), increased (102,136) as well as unaltered (137,138) motor thresholds, thus yielding inconclusive evidence for hyper- or hypo-excitability in this region (89). Combining TMS with EEG (139) could provide direct and objective markers of cortical responsivity including changes in network excitation or inhibition as shown for studies in epilepsy (140,141) but not yet applied in migraine.
Cortical neurophysiological changes during the premonitory, aura and ictal phase
Also, no definite EEG abnormalities have been consistently identified during migraine attacks (86,96). Decreased alpha band power and asymmetry with respect to the painful side were observed prior to and during attacks without aura compared to the interictal phase (142), in line with alpha band asymmetry reported prior to migraine attacks with aura (143). Longitudinal recordings revealed preictal changes including EEG slowing, alpha and theta band asymmetry in occipito-parietal and temporal regions (97,144,145), as well as enhanced EEG power (145,146) and coherence (97,144,146). In the study by Cao et al., EEG power and coherence were reduced in interictal and ictal phases with respect to healthy controls, suggesting “normalization” of network changes preictally (146). EEG features discriminating attacks with versus without aura have not been identified yet (86,88,97,144). In addition to common migraine, EEG studies in hemiplegic migraine are of particular interest given translational insight from FHM mouse models (48). While early studies report no effect of hemiplegia on EEG lateralization (147,148), a patient with sporadic hemiplegic migraine and SCN1A mutation displayed slow waves spreading from posterior to anterior cortical regions during a hemiplegic attack (149). Carriers of the FHM1 S218L mutation in the CACNA1A gene, which causes a severe neurological phenotype of hemiplegic migraine with possible seizures, can display epileptiform EEG abnormalities outside attacks (150).
For pattern reversal VEPs, amplitude features were enhanced prior to attacks (113,114), in particular for high contrast and spatial frequency stimuli, suggesting a cyclic reduction in intracortical inhibition of extra-striatal regions (18 and 19) of the occipital cortex (114). Habituation of evoked responses was described to be either impaired or unaltered between attacks and comparable to controls during attacks for visual and several other stimulation modalities (90,93). Since longitudinal designs were only used in studies that did not observe interictal lack-of-habituation (108,114), it remains to be seen whether cyclic changes in evoked response habituation (120) actually occur. Other observations nevertheless support the idea that transient changes in visual cortex responsivity occur prior to attacks, since photic drive responses (reported to be enhanced interictally) showed attenuation in the preictal period (97). For the motor cortex, responses to short-burst repetitive TMS differed over the migraine cycle for migraine with and without aura (90), suggesting attack-related changes in excitability. In line with this view, motor thresholds to single pulse TMS were higher for shorter time-intervals from the last attack (151).
With respect to the migraine aura, absence of EEG changes during CSD could be explained by filtering of slow potential changes with standard clinical EEG or may simply reflect difficulties in detecting CSD through the intact scalp (152–155). Observations from patients with ischemic brain injury showed that CSDs can manifest in scalp EEG as depressions of ongoing activity, with identification of such depressions not being straightforward (155,156). In fact, there is currently a lack of criteria to identify CSD based on EEG alone, which may be related to limitations of data analysis methods used, true lack of CSD in EEG recordings or low CSD incidence in the study population, among other shortcomings. Advanced signal processing to identify a possible EEG signature of CSD may be needed to improve our understanding of CSD in future studies. To make it even more complicated, auras may be caused by heterogeneous and spatially restricted CSD events as directly visualized in the human cortex using intrinsic optical signal and cerebral blood flow monitoring (157,158). These locally restricted CSDs might only cause local neuronal network depression in narrow parts of the cortex (152) that cannot be detected by standard EEG. MEG can solve at least some of these issues by its ability to detect CSD based on magnetic fields arising from the synchronous and spreading depolarization of cortical neurons (159,160). Indeed, using MEG, a spreading depolarization-like neuroelectric event (161) as well as alpha band desynchronization in the left extra-striate occipital and temporal cortex (162) were associated with visual aura symptoms, while gamma frequency desynchronization occurred during sustained inhibition of visual function after cessation of aura (162).
Preclinical studies
Animal models are valuable tools to explore mechanisms of cortical dysfunction in the context of migraine. Translational approaches include introduction of human pathogenetic mutations, like those for FHM or casein kinase 1 delta (163), chemical, mechanical or optogenetic CSD induction (164,165), and/or chemical sensitization of trigeminovascular pathways using, for example, nitroglycerin (166) or CGRP (167). FHM1 mouse models are well characterized at both cellular and functional network levels, yielding insight into the role of glutamatergic excitatory mechanisms in hemiplegic migraine, and to a certain extent also common forms of migraine (48,168). Cortical plasticity has been implicated by some of the structural and functional clinical studies to contribute to attack recurrence and chronification (11,32) and can be investigated mechanistically using hypothesis-driven experiments in rodents. Figure 4 summarizes the key preclinical data on cortical structure and function in migraine, outlined in detail below.
Schematic representation of the rodent brain with indication of the primary visual cortex V1 and primary somatosensory cortex S1, in close connection to thalamic nuclei (Th) that are part of the trigeminovascular pain system. See main text for details.
CSD studies in migraine animal models
CSD has been widely studied in animal models as the electrophysiological correlate of the aura phase. In addition, CSD is a putative headache trigger based on experimental findings that CSD can activate brainstem and other regions involved in headache mechanisms (169–172), although clinical evidence is lacking and the occurrence of spreading depolarization could be debated for migraine attacks not preceded by aura (173,174).
FHM1 mice displaying the gain-of-function missense mutations R192Q or S218L in the Cacna1a gene encoding a subunit of neuronal voltage-gated CaV2.1 Ca2+ channels show enhanced CSD susceptibility (175–177), particularly in the severe S218L mutant (175,176). Heterozygous FHM2 knock-in mice expressing the loss-of-function W887R missense mutation (178,179) in the Atp1a2 gene, encoding the α2 subunit of the Na+/K+ pump ATPase present on astrocytes, and mice carrying the missense mutation T44A in the casein kinase Iδ gene involved in circadian rhythmicity (180) also exhibit an increased CSD susceptibility.
Cortical network changes underlying enhanced CSD susceptibility in a migraine-susceptible brain have been studied in most detail in the FHM1 models. Ca2+ influx appears enhanced in cortical slices of FHM1 R192Q and S218L mice as a direct consequence of hyperactive CaV2.1 channels (181–183). Cortical in vivo Ca2+ imaging provided further evidence for a facilitated cortical resting state in heterozygous S218L mice. Elevated neuronal Ca2+ level at rest was associated with altered synaptic morphology compatible with stronger synapses and a hyperexcitability phenotype (184). For FHM2, the enhanced CSD susceptibility in heterozygous mutants was found to be associated with impaired K+ and glutamate clearance by cortical astrocytes in the somatosensory cortex (179).
Translational value of CSD readouts is underscored by the observation that the same factors modulate susceptibility to CSD and migraine. For example, female FHM1 mice are more susceptible to CSD than males, in line with a female preponderance in migraine (185). This sex difference could be abrogated by hormonal ablation experiments in FHM1 mice (175,185), suggesting potentiating effects of sex hormones on cortical excitability in a genetically susceptible brain. Similarly, stress hormone corticosterone pre-treatment enhanced CSD susceptibility specifically in FHM1, but not in wild-type mice (186). In addition, prophylactic migraine drugs (187,188), as well as non-invasive neuromodulation by TMS (189) or vagus nerve stimulation (190), suppress CSD susceptibility or propagation in rodents. With respect to effects of endogenous neuromodulators, CGRP was found to be released during CSD in rodent brain slices in vitro (191). CGRP receptor antagonists inhibit CSD in vitro (191) and CSD-related hyperemia in vivo (192), while aborting migraine attacks in patients (193). In addition to receptor antagonists, CGRP receptor antibodies have shown potential in attack prevention (194).
Insight into molecular pain-related mechanisms following CSD came from the observation that induction of CSD events in wild-type mice activates neuronal Pannexin channels that initiate an inflammatory cascade and subsequent activation of the trigeminovascular system (172). Accordingly, inhibition of Pannexin1 mega channels suppresses CSD (195). A recent paper demonstrated that, following CSD induction, dynamic activation and migration of macrophages and dendritic immune cells mediates both acute and delayed activation of meningeal nociceptors, as a mechanism for CSD activating headache both directly and with a delay (196). It is possible that changes in the cortex of migraine patients may predispose to such inflammatory changes, as indicated by specifically altered inflammatory profiles following CSD in FHM1 mutant mice (197).
Cortical plasticity changes in migraine models
Cortical network plasticity has been studied less than mechanisms of CSD in migraine models. At the morphological level, FHM1 mice display altered axonal and dendritic morphology in the sensorimotor cortex, with larger axonal boutons and a higher percentage of highly excitable mushroom-type dendritic spines, which are densely populated with excitatory NMDA receptors compared to wild-type, suggesting stronger and more excitable synapses (184). This is in line with differentially expressed proteins involved in neurite outgrowth and actin dynamics in cortical synaptic proteomes from FHM1 mice (198). Some light on cortex-specific changes in migraine was shed by observations that functional effects of FHM1 mutations appear neuron type-specific. Comparison of cortical and brainstem neurons revealed that only cortical pyramidal neurons exhibit enhanced Ca2+ influx as result of the mutation, which was related to the typical long-duration/small amplitude shapes of action potentials in these cells (199). Further, FHM1 mutations do not exert an effect on fast-spiking cortical interneurons, which contrasts the clear gain-of-function effect observed for excitatory pyramidal neurons (181,182). Effects of FHM1 mutations across cortical (as well as other) regions therefore may be context-dependent and result in dynamic disturbances of the balance between excitation and inhibition in neuronal circuits (200).
Functional consequences of CSD on cortical networks have been investigated as well. Evidence for cortical synaptic potentiation following CSD comes from a study in freely behaving rats, in which cortico-cortical evoked responses and brain derived neurotrophic factor were increased in the somatosensory cortex directly after a 1-hour period of local CSD induction, in the CSD-affected cortical hemisphere (201). Under anesthesia, recovery of whisker-evoked response in the somatosensory cortex following CSD did not show potentiation in wild-type or FHM1 mutant mice (175); in contrast, evoked responses to fore- and hindpaw stimulation showed variable changes in rats indicative of CSD-induced cortical plasticity (202). Repeated daily CSD induction in the frontal cortex causes reactive astrocytosis associated with reduced susceptibility to CSD (203). When studying effects of induced CSD, one should take into account possible heterogenous spreading patterns, as revealed by wide-field imaging (204) and in line with clinical descriptions of variable aura patterns in patients (157) that may cause complex functional and molecular changes.
Taken together, the observations above show that CSD impacts cortical network plasticity, which may be even more pronounced under freely behaving conditions, with possible implications for mechanisms underlying attack recurrence and chronification (11,32).
Future strategies for investigating cortical structure and function in migraine
Considering the range of data on cortical morphology and function in migraine, only a few clinical studies have used longitudinal designs, and most preclinical studies were performed under anesthesia, in terminal experiments. Having a migraine patient or an animal serve as its own control is crucial to identify cyclic changes in brain activity that precede and/or influence attack initiation and recurrence. Such information carries quantifiable measures that can indicate (i) worsening of disease state, (ii) transitioning from normal physiology to pathological activity, and (iii) treatment efficacy. Identification of baseline biomarkers that predict and monitor treatment response in migraineurs would provide important tools for personalized medicine. Logistically, repeated neuroimaging (59) poses a larger challenge than longitudinal EEG. Technological developments in ambulatory EEG devices can advance the field by allowing brain monitoring for prolonged periods of time, in a patient's natural habitat, in particular when including systems physiology data (e.g. blood pressure, heart rate, body temperature, movement) using smart sensors. In preclinical research, cellular-resolution neuroimaging (205) and combined cortical and brainstem network neurophysiology in freely behaving animals (206) can help uncover cellular and network features underlying migraine susceptibility and bridge gaps between clinical neuroimaging and EEG. Non-invasive modulation of brain activity and induction of CSD by optogenetics (165,207), TMS (189) or ultrasound (208) provide further translational advancement by reducing confounding effects of invasive surgery on cortical function. Longitudinal designs under awake conditions allow certain drugs, neuromodulation or trigger paradigms to be tested on different readouts (e.g. EEG features, CSD, allodynia, pain thresholds) in the same animal at repeated moments in time. While already implemented in epilepsy (209), it is expected that also for understanding and normalizing cortical dysfunction in migraine, differentiation of human-induced pluripotent stem cells into 2D and 3D models provides novel opportunities for investigating (personalized) effects of treatments and triggers on cortical neurons differentiated from patient-derived stem cells.
Conclusions
An important role of the cortex in migraine is well acknowledged for the initiation of aura via the electrophysiological phenomenon CSD, and more recently also for the perception of head pain and associated sensations. Structural and functional findings suggest that static and plastic changes within and across cortical regions contribute to enhanced migraine susceptibility and its clinical course by modulating trigeminovascular pain and sensory processing networks.
It is important to acknowledge that clinical morphometric and functional neuroimaging data, as well as neurophysiological data on cortical function, show partially contradicting results. This could relate to shortcomings with respect to: (a) the relatively low number of patients in many studies; (b) there being only a few replication studies given the complexity of protocols and differences in equipment; (c) differences in analyses (e.g. whole-brain imaging vs. selective brain regions, or low-density EEG); (d) variation in methods of data collection; (e) differences in timing of data collection with respect to migraine attack onset; (f) differences in medications and comorbidities that often are not taken into account; (g) study design – longitudinal studies are needed to evaluate how a change in clinical phenotype affects imaging or neurophysiological findings. These methodological differences between studies make meta-analysis difficult. In addition, it remains unclear whether positive findings are consequences and/or causes of migraine. Hypothesis-driven designs should take into account the interplay between cortical regions and subcortical structures such as the brainstem, thalamus and hypothalamus, as well as the static and cyclic effects of internal (e.g. genetics, gender, metabolism) as well as external factors (e.g. stressors, circadian and environmental changes) on these processes.
Footnotes
Clinical implications
Structural and functional changes across cortical regions in migraineurs are evidenced by altered grey and white matter volumes as well as neuronal network and function.
These changes likely contribute to initiation, cyclic recurrence and chronification of attacks.
Morphometric and functional imaging, as well as neurophysiology, can provide diagnostic tools to help distinguish different migraine subtypes from each other, including chronic from episodic migraine, and thereby help in guiding therapy.
Preclinical studies revealed a critical role for cortical hyperexcitability in enhancing CSD susceptibility, thereby helping to identify treatment targets to prevent attack initiation and chronification.
Declaration of conflicting interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.