
Abstract
Background
Migraine aura reflects transient neurological disturbances attributed to cortical spreading depolarization (CSD), yet the upstream molecular events that promote or precipitate this phenomenon remain uncertain. Human pharmacological provocation models provide a controlled approach to identifying endogenous signaling pathways capable of triggering aura and thereby clarifying their mechanistic role in migraine pathogenesis.
Methods
This systematic review was pre-registered in PROSPERO (ID: CRD420250636266) and conducted in accordance with the Synthesis Without Meta-Analysis (SWiM) guideline. PubMed and Embase were searched from database inception through January 1, 2026, without language restrictions, to identify experimental studies administering pharmacological agents to individuals with migraine with aura under controlled lab conditions. Two reviewers independently performed study selection, data extraction, and methodological assessment. Extracted variables included participant characteristics, study design, pharmacological trigger, dosing protocol, and the incidence, timing, and phenotype of provoked aura and headache. Because of substantial heterogeneity, findings were synthesized qualitatively.
Results
Fourteen studies met inclusion criteria, examining seven pharmacological agents: calcitonin gene-related peptide (CGRP), levcromakalim (ATP-sensitive potassium (KATP) channels opener), glyceryl trinitrate (GTN), sildenafil, cilostazol, endothelin-1, and histamine. Across studies, aura induction occurred far less frequently than headache induction. CGRP provoked aura in 17 (32%) of 53 participants across three studies, with latencies ranging from 10 to 360 min. Pharmacological opening of vascular KATP channels by levcromakalim elicited aura in 14 (27%) of 52 participants in randomized crossover trials, with onset between 20 and 120 min. Other agents produced minimal or no aura responses.
Conclusions
Human pharmacological provocation provides a reproducible framework for dissecting molecular pathways that can trigger migraine aura. Current evidence implicates CGRP signaling and vascular ATP-sensitive potassium channel activation as the most plausible associated candidate pathways. However, standardized protocols and larger controlled studies are required to confirm these mechanisms and refine experimental models of aura biology.
Trial Registration
PROSPERO (ID: CRD420250636266).
This is a visual representation of the abstract.
Introduction
Migraine with aura is characterized by transient neurological disturbances that develop over minutes and reflect the propagation of cortical spreading depolarization (CSD).1,2 This slow wave of neuronal and glial depolarization travels at 2 to 5 mm per minute, presumably creating the spatial gradient that shapes aura progression. 3 As the wavefront advances, it disrupts cortical signaling, and its passage is followed by transient suppression of neural activity, which are thought to correspond to the nature of transient positive and negative neurological disturbances reported during aura. 4 These linked processes produce the characteristic temporal and spatial evolution of spontaneous aura.
CSD also initiates downstream events relevant to migraine pathogenesis. Preclinical studies show that it activates the trigeminovascular system, promotes the release of pro-inflammatory mediators, and induces meningeal vasodilation, suggesting a biological bridge between aura and migraine headache.5–8 However, the physiological conditions that permit or trigger CSD in humans remain uncertain. This uncertainty limits understanding of why aura precedes, follows, or overlaps with headache in individual attacks. As a result, the temporal relationship between these phenomena remains incompletely defined, with evidence supporting sequential, bidirectional, or parallel interactions.9–12
Progress in clarifying these interactions requires direct observation of aura physiology, yet spontaneous aura rarely occurs under conditions that allow standardized study. Its brief duration and unpredictable onset restrict opportunities for systematic measurement in clinical or research settings. 13 Human provocation models were developed to address this challenge by enabling safe and reproducible induction of migraine aura in controlled laboratory settings.14,15 Early protocols focused on provoking migraine headache alone, but recent approaches now test whether pharmacological agents can also trigger aura in participants with migraine.14,16–18 This shift reflects increasing interest in identifying molecular mechanisms that initiate CSD or reduce the threshold for its occurrence.
Pharmacological provocation supports this effort by providing a controlled platform for mechanistic investigation. 15 It enables precise characterization of temporal relationships between agent exposure and symptom onset, offering insights unavailable in spontaneous attacks. It also allows structured comparison across distinct molecular triggers, thereby revealing pathway-specific effects on cortical excitability. Interestingly, non-pharmacological stimuli such as hypoxia, visual stimulation, or carotid puncture can induce aura, but their mechanisms are diffuse and difficult to isolate.19,20 Pharmacological agents, in contrast, engage defined biological pathways with reproducible dosing. 21 Nonetheless, consistent aura induction remains uncommon, and existing studies vary in design, participant selection, and outcome reporting, complicating interpretation.
These limitations underscore the need for a comprehensive synthesis of existing evidence. This systematic review integrates all published human provocation studies assessing aura induction in participants with migraine with aura, summarizes their principal findings, and evaluates their implications for understanding migraine aura biology.
Methods
This systematic review was pre-registered in the International Prospective Register of Systematic Reviews (PROSPERO, CRD 420250636266) and conducted in accordance with the Synthesis Without Meta-analysis (SWiM) guideline. 22
Overview of the human provocation model
The human provocation model provides a reproducible and controlled framework for investigating migraine mechanisms under lab conditions. 21 It is founded on the principle that administering signaling molecules capable of inducing migraine attacks in persons with migraine can reveal molecular mechanisms central to disease pathogenesis. 16 The reversible and treatable nature of migraine attacks allow these responses to be studied safely in lab settings. Initially developed to examine migraine headache, the model has been refined to determine whether pharmacological agents can also trigger migraine aura in individuals diagnosed with migraine with aura.14,17 This adaptation enables systematic evaluation of aura attacks that are otherwise sporadic and short-lasting, thereby facilitating direct observation of its biological correlates.
Experimental protocols most often apply randomized, double-blind, placebo-controlled crossover designs, although open-label studies have been conducted in exploratory contexts. Participants must be free from both aura and headache and abstain from acute medications for at least 24 to 48 h before provocation.17,23
Baseline evaluations include vital parameters, aura and headache status, and accompanying symptoms. 17 Following intravenous or oral administration of the provoking agent, participants are observed at regular intervals—typically every ten minutes—for 1 to 2 h and continue symptom recording on an hourly basis for up to 12 h after exposure.
An experimentally induced migraine headache is defined as an attack fulfilling International Classification of Headache Disorders (ICHD-3) criteria C and D for migraine without aura, or one replicating the participant's usual spontaneous migraine headache. 24 An experimentally induced aura is defined as the occurrence of neurological symptoms meeting ICHD criteria B and C for migraine with aura. 25
Search strategy
A systematic search of PubMed and Embase was conducted to identify clinical investigations using the human provocation model to study the induction of migraine aura. The search encompassed all available records from database inception through January 1, 2026, without language restrictions. No publication date restrictions were applied. Both free-text keywords and Medical Subject Headings (MeSH) terms were combined to capture studies addressing migraine with aura and pharmacological provocation paradigms. The complete PubMed and Embase search strings are provided in Supplemental Table 1. In addition, the reference lists of all included studies and relevant systematic reviews were examined manually to ensure coverage of the available evidence.
Selection criteria
Studies were eligible for inclusion only if they enrolled participants with a confirmed diagnosis of migraine with aura according to ICHD criteria and prospectively evaluated aura occurrence following administration of a defined pharmacological trigger under controlled lab conditions.
Two independent reviewers (ZH and BAC) systematically screened all titles and abstracts to determine potential eligibility. Full-text articles were retrieved for studies meeting inclusion criteria or for which eligibility could not be confirmed from the abstract. Disagreements between reviewers were resolved through discussion or, when necessary, consultation with a third reviewer (HMA). The overall selection process is illustrated in the Preferred Reporting Items for Systematic reviews and Meta-Analyses (PRISMA) flow diagram (Figure 1), and detailed eligibility criteria are provided in Supplemental Table 2.

Study flow diagram.
Data extraction
Data were independently extracted by two reviewers (ZH and BAC) using a pre-specified standardized form. Extracted variables encompassed study-level characteristics, including authorship, publication year, and design, as well as participant demographics such as sample size, age, and sex. Information regarding the molecular triggers used was recorded in detail, including compound type, administered dose, and route of administration. For each provocation study, data on headache induction were collected, encompassing attack intensity, quality, localization, associated symptoms, duration, and time to onset. Aura-related variables were similarly extracted, including subtype, timing, anatomical distribution, duration, and detailed symptom descriptions. Discrepancies between reviewers were resolved through discussion and, when necessary, adjudicated by a third reviewer (HMA).
Data synthesis
Because of substantial heterogeneity across study designs, participant populations, and intervention protocols, a quantitative meta-analysis was not feasible. Instead, a structured qualitative synthesis was performed, integrating study characteristics, methodological parameters, and primary findings into both tabular and narrative formats. To facilitate interpretability, studies were organized according to the pharmacological trigger investigated.
Risk of bias was independently assessed by two reviewers in accordance with the Cochrane Handbook for Systematic Reviews of Interventions, evaluating random sequence generation, allocation concealment, blinding, incomplete outcome data, selective reporting, and other sources of bias. 26
Results
A total of 2007 records were identified across PubMed and Embase. After removing 463 duplicates, 1544 titles and abstracts were screened. Following full-text review and manual reference searches, 14 studies fulfilled the eligibility criteria and were included in the qualitative synthesis18,25,27–38 (Figure 1). The included studies were published between 1999 and 2024.
Sample sizes for participants diagnosed with migraine with aura ranged from 3 to 34 (Figure 2). Thirteen studies examined aura induction in individuals with migraine with aura accompanied by headache,18,25,27–33,35–38 and one enrolled participant who experienced aura without headache. 34 Six studies applied randomized, double-blind, placebo-controlled crossover designs,32–37 whereas six were open-label,18,25,27,28,30,31 one was double-blind, 38 and one was single-blind. 29 Overall, double-blind randomized studies were judged at low risk of selection, performance, and detection bias, whereas open-label or uncontrolled studies were judged at higher risk (Supplementary Table 3).

Aura induction rates across pharmacological triggers. GTN: glyceryl trinitrate, CGRP: calcitonin gene related peptide, ET-1: endothelin-1, n: number of participants, %: percentage.
Across these 14 studies, seven pharmacological agents were investigated for their ability to trigger migraine aura: glyceryl trinitrate (GTN) (n = 4),18,25,27,28 calcitonin gene-related peptide (CGRP) (n = 3),29–31 levcromakalim (n = 3),32–34 cilostazol (n = 1), 35 sildenafil (n = 1), 36 endothelin-1 (n = 1), 37 and histamine (n = 1). 38 Intravenous infusion represented the most frequent route of administration (n = 10),18,27–34,37 followed by oral (n = 2),35,36 sublingual (n = 1), 25 and inhalational (n = 1) delivery. 38 All studies used diagnostic criteria from the ICHD: three applied ICHD-1,18,25,38 two ICHD-2,27,29 four ICHD-3β,28,35–37 and five ICHD-3 criteria.30–34
Medication policies varied across studies. Two allowed continuation of stable preventive therapy,30,31 seven required no preventive medication use at the time of participation,18,25,27,32,33,35,36 and five did not specify medication status.28,29,34,37,38 General study characteristics and principal findings are summarized in Table 1–3.
Characteristics and outcomes of pharmacological provocation studies in migraine with aura using nitric oxide donors, phosphodiesterase inhibitors, endothelin-1, and histamine.

GTN: glyceryl trinitrate, ET-1: endothelin-1, NA: not applicable, IV: intravenous, n: number, %: percentage, min: minutes, RCT: randomized placebo-controlled trial, ICHD: international classification of headache disorders, μg: microgram, mg: milligram, kg: kilogram, ml: milliliter.
*Defined criteria applied to induced aura episodes.
Characteristics and outcomes of pharmacological provocation studies in migraine with aura using calcitonin gene-related peptide.
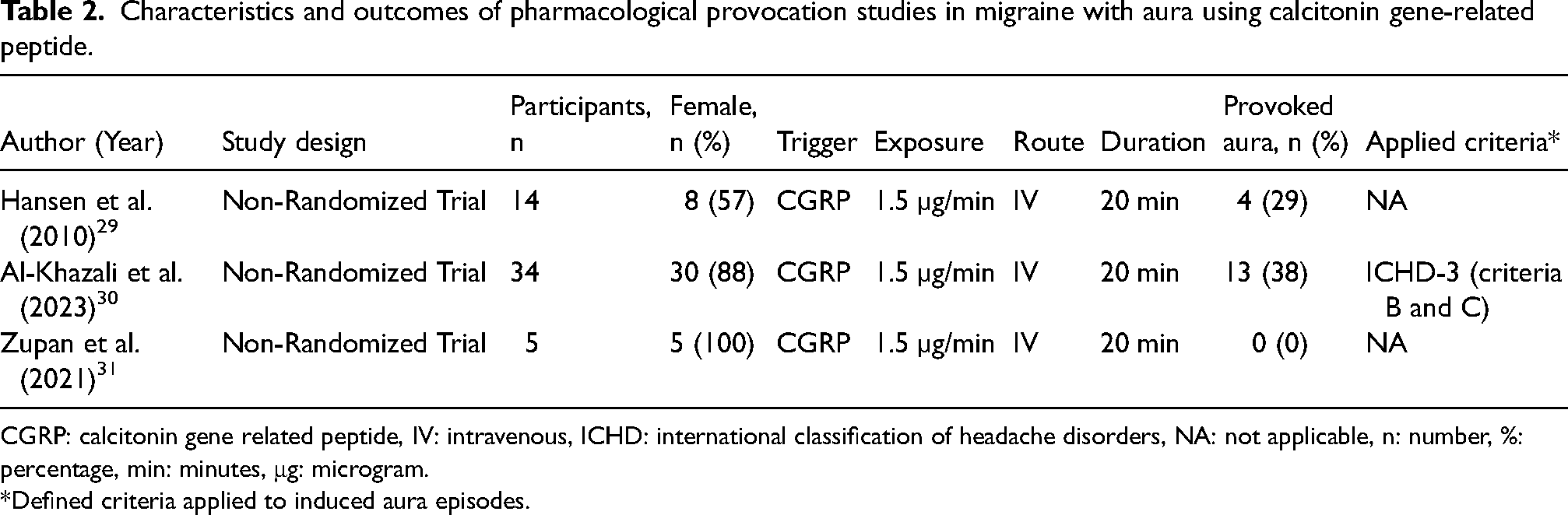
CGRP: calcitonin gene related peptide, IV: intravenous, ICHD: international classification of headache disorders, NA: not applicable, n: number, %: percentage, min: minutes, μg: microgram.
*Defined criteria applied to induced aura episodes.
Characteristics and outcomes of pharmacological provocation studies in migraine with aura using levcromakalim.

IV: intravenous, NA: not applicable, n: number, %: percentage, min: minutes, RCT: randomized placebo-controlled trial, ICHD: international classification of headache disorders, mg: milligram.
*Defined criteria applied to induced aura episodes.
GTN
Four studies investigated GTN as a pharmacological provocation agent,18,25,27,28 three of which included healthy controls (HCs).18,25,27 Intravenous infusion administered over 20 min was used in three studies,18,27,28 whereas one used sublingual delivery. 25 Across these investigations, aura induction was uncommon, whereas migraine headache was consistently provoked (Table 1).
In the first non-randomized, open-label study, none of the 12 participants with migraine with aura developed aura following intravenous infusion, although migraine headache occurred in six (50%). 18 None of the 14 HCs experienced either aura or headache. A subsequent non-randomized, open-label used the same infusion protocol in 21 participants with migraine with aura and 11 HCs to examine reproducibility across two sessions. 27 Migraine headache developed in 14 (67%) participants with migraine with aura during the first session. A single participant experienced a visual aura after the first infusion and again during the second, with similar onset times of 150 and 160 min. One additional participant developed visual aura during the second session, while no aura or headache occurred among HCs. Among 23 participants with migraine without aura, 19 (83%) developed a migraine headache following the infusion.
A third non-randomized, single arm, open-label investigation by the same research group evaluated reproducibility in 32 participants with migraine with aura and 21 with migraine without aura. 28 During the first session, seven (22%) participants with migraine with aura experienced aura. In the second session, which included 15 participants, aura recurred in four participants (27%). The report did not specify whether aura occurred exclusively among those with a confirmed history of aura, nor were migraine headache rates or aura characteristics detailed.
The final non-randomized, open-label study administered sublingual GTN to 22 participants with migraine with aura, 168 with migraine without aura, and 53 HCs. 25 Aura developed in three (14%) participants with migraine with aura, each describing transient visual disturbances (e.g., phosphenes, scotomas) resembling their typical spontaneous aura. Migraine headache followed in seven (32%) participants with migraine with aura, and 138 (82%) participants with migraine without aura.
CGRP
Three studies investigated human α-CGRP's potential to induce migraine aura, all using intravenous infusion at 1.5 µg per minute for 20 min.29–31 One included a HCs, 29 whereas the remaining two were open label.30,31 (Table 2)
In the first non-randomized, single-blind study, 14 participants diagnosed with migraine with aura and 11 HCs received intravenous CGRP. 29 Migraine aura developed in four participants (29%), including one prolonged episode lasting 10 h. The median onset of aura symptoms was 70 min (range, 10–360 min). Reported phenomena included fortification spectra, bilateral flickering lights, left-sided homonymous scotoma, and tingling paresthesia. Migraine headache followed in eight participants (57%), whereas none of the HCs experienced migraine aura.
A second single-arm, open-label study involving 34 participants with both migraine with and without aura found that 13 (38%) reported aura within 12 h after infusion. 30 The median onset was 30 min (range, 10–360 min). Visual aura occurred in 10 participants and somatosensory aura in 7, several experiencing both. In 11 of 13 cases, migraine aura appeared after headache onset, and all described the provoked symptoms as closely resembling their spontaneous aura. Migraine headache occurred in 24 participants (71%), with a median onset of 40 min (range, 10–240 min). The authors found that those who experienced migraine aura after CGRP infusion had a higher frequency of migraine aura in the past year, compared with those who did not.
A third single-arm, open-label study examined cerebral hemodynamic responses to CGRP in 5 participants with migraine with aura, and 15 with migraine without aura. 31 None of participants with migraine with aura experienced aura, although all developed migraine headache. Among participants with migraine without aura, 11 (73%) developed a migraine headache following the infusion.
Levcromakalim
Three randomized, placebo-controlled crossover trials assessed levcromakalim's ability to induce migraine aura using a standardized 20-min intravenous infusion32–34 (Table 3).
In the first study, 17 participants with migraine with aura received both levcromakalim and placebo on separate days. 32 Aura occurred in 10 participants (59%) after levcromakalim, with a median onset of 44 min (range, 20–120 min). All aura attacks involved visual disturbances such as flickering lights, blurred vision, dark spots, or scotomas, while 3 participants also experienced somatosensory symptoms including paresthesia or numbness. Migraine headache developed in 14 participants (82%) after levcromakalim, with a median onset of 168 min (range, 60–240 min). No aura or headache occurred following placebo.
A second trial enrolled 27 participants who had a diagnosis of both migraine with and without aura. 33 Following intravenous infusion of levcromakalim, four (15%) participants reported experiencing migraine aura, whereas two (7%) did so after placebo (a non-significant difference). The median onset of aura symptoms was 27.5 min (range, 30–323 min). Reported aura symptoms included visual phenomena (n = 3), somatosensory disturbances (n = 2), and subjective motor symptoms (n = 1). Migraine headache developed in 14 (52%) following levcromakalim compared with three (11%) after placebo, with a median latency of 180 min (range, 30–360 min).
A third examined eight individuals who experienced aura without headache. 34 None developed aura after levcromakalim, whereas two (25%) reported aura after placebo. Migraine-like headache occurred in five (63%) following levcromakalim.
Cilostazol
A randomized, placebo-controlled crossover study evaluated the effects of oral cilostazol in 16 participants diagnosed with migraine with and without aura. 35 Migraine aura occurred in one (6%) participant after cilostazol and in another (6%) after placebo. The cilostazol-induced aura manifested as a left-sided homonymous scotoma beginning 4.5 h post-ingestion, whereas the placebo-related aura appeared 15 min after administration. Migraine headache developed in 12 (75%) participants after cilostazol and in one (6%) after placebo, with a median onset of 330 min (range, 120–600 min).
Sildenafil
A randomized, placebo-controlled crossover study evaluated the ability of oral sildenafil to provoke migraine aura in 16 participants diagnosed with both migraine with and without aura. 36 Migraine aura developed in three (19%) participants after sildenafil administration but in none after placebo. The median time to aura onset was 150 min (range, 60–180 min). Two participants reported unilateral flickering light spots, and one described bilateral scintillating phenomena, all resembling their typical spontaneous aura attacks. Migraine headache occurred in nine participants (56%) after sildenafil, with a median onset of 300 min (range, 120–360 min), whereas no headaches were reported after placebo.
Endothelin-1
A randomized, placebo-controlled crossover study evaluated the ability of endothelin-1 to induce migraine aura through a 20-min intravenous infusion administered to 14 participants diagnosed with migraine with and without aura. 37 None of the participants developed aura on either experimental day, and migraine headache occurred in only one individual (7%) following endothelin-1 infusion, with onset approximately 120 min post-administration.
Histamine
A single non-randomized, double-blind study investigated the effects of histamine inhalation in three participants diagnosed with migraine with aura, 12 with migraine without aura, and 15 HCs. 38 All participants received escalating doses of inhaled histamine under controlled lab. None of the participants with migraine with aura or the HCs developed aura following exposure. Headache outcomes were not reported for the migraine group.
Discussion
This systematic review synthesized all available human pharmacological provocation studies evaluating whether defined molecular pathways can induce migraine aura under controlled laboratory conditions. Fourteen studies met inclusion criteria and collectively examined seven endogenous or vasoactive compounds. Across these investigations, aura induction occurred far less often than migraine headache induction, underscoring that the two phenomena, although clinically linked, are not mechanistically identical. Importantly, negative provocation findings should not be interpreted as evidence against biological involvement of a given pathway in aura pathogenesis. They might instead reflect limitations in model sensitivity, including dosing paradigms, timing, observation windows, outcome definitions, and study design.
Of all tested triggers, CGRP and the vascular KATP channel opener levcromakalim were associated with the induction of aura. However, aura induction rates remained modest, and these findings should be interpreted as evidence of association rather than definitive mechanistic proof. About one-third of participants receiving CGRP and one quarter receiving levcromakalim developed aura, whereas no aura occurred in HCs or in participants with migraine without aura. Across studies, provoked aura symptoms closely resembled spontaneous aura, lending support to the validity of the provocation paradigm. Together, these findings identify CGRP and vascular KATP activation as the most promising candidate pathways for experimental induction of migraine aura.
Existing evidence and added value
The findings of this review build on several decades of work defining the biological correlates of aura and its relationship to migraine headache. Early experimental studies demonstrated that CSD produces a wave of oligemia and altered neurovascular coupling that mirrors the clinical evolution of aura. 39 Subsequent research showed that CSD releases potassium, hydrogen ions, and vasoactive peptides capable of activating perivascular trigeminal afferents.6,39,40 More recent work suggests that CSD might also influence trigeminovascular activation through glymphatic-mediated cerebrospinal fluid signaling from the cortex to the trigeminal ganglion. 41 These observations established a physiological bridge between cortical events and trigeminovascular activation, yet they did not clarify which certain signaling pathways promote CSD initiation in humans.
Human provocation models help address this gap by testing whether well-defined molecular triggers can elicit migraine aura under controlled experimental conditions. The present synthesis advances the field by assessing such triggers across studies, demonstrating that intravenous CGRP administration and vascular KATP channel activation show the highest reproducibility in eliciting aura. These findings across independent investigations suggests that certain signaling pathways exert direct or indirect modulatory effects on cortical excitability, even though their primary sites of action are located outside the central nervous system (CNS). 42 Notably, the potential of other neuropeptides such as pituitary adenylate cyclase-activating polypeptide (PACAP) to elicit aura remains unexplored, despite their established ability to trigger migraine headache, representing an important gap for future research. 43
This review further underscores the importance of individual susceptibility, particularly baseline aura frequency, as a potential key determinant of responsiveness to experimental provocation. 30 In addition, the experimental evidence highlights important limitations in using headache induction alone as a surrogate marker for CSD susceptibility, as molecular pathways that reliably provoke headache do not necessarily trigger aura. This dissociation might, at least in part, reflect dose–response effects, whereby higher stimulus intensities or doses are required to exceed the threshold for aura generation, whereas lower levels of activation can be sufficient to elicit migraine headache without aura. Taken together, these insights emphasize the need for more targeted and mechanistically informed provocation paradigms capable of testing hypotheses about migraine aura with greater precision.
Hypothesized mechanisms
The mechanistic implications of these findings can be interpreted within two hypotheses describing the relationship between aura and headache: the sequential or build-up model and the bidirectional neurovascular model. Evidence from this review, together with findings from preventive migraine treatment, places these models in clearer perspective.
The sequential model proposes that CSD is the primary initiating event in migraine with aura.5,7 In this model, CSD disrupts cortical homeostasis and releases potassium, glutamate, hydrogen ions, and pro-inflammatory mediators into the cerebrospinal fluid. 5 These mediators then activate meningeal nociceptors and initiate trigeminovascular signaling, producing headache once a critical threshold is reached. The build-up hypothesis refines this model by linking headache timing to the cumulative concentration of these mediators, thereby explaining variability in the latency between aura and headache. 44
Extensive preclinical work supports this directionality. CSD consistently activates meningeal afferents, increases cellular FOS (c-Fos) expression in trigeminovascular nuclei, and produces headache-like behaviors in rodents. 45 These findings make a strong case that CSD leads totrigeminovascular activation is a robust biological pathway.
However, the sequential model alone cannot explain key clinical observations. Foremost among them is the consistent preventive effect of peripherally restricted anti-CGRP monoclonal antibodies on both migraine headache and seemingly also migraine aura.46–48 These antibodies do not cross the blood–brain barrier in meaningful quantities, yet some interventional studies report reductions in aura frequency. A purely sequential model would require that aura prevention occur through direct cortical effects of treatment, which is incompatible with the pharmacology of these antibodies. This discrepancy suggests that a peripheral pathway capable of modulating CSD susceptibility must exist, and that this pathway is not captured by the traditional sequential view.
The bidirectional model addresses this limitation by proposing that peripheral trigeminovascular activation can influence cortical excitability and promote CSD initiation.30,49 This 2023 hypothesis is supported by human provocation studies summarized in this review. In this model, activation of meningeal nociceptors produces ascending signals through the trigeminal pain pathways. These inputs modulate thalamocortical circuits projecting to cortical regions, including the visual cortex, where CSD is most commonly initiated. 50 In people with migraine with aura, this modulation might be sufficient to lower the CSD threshold.
The bidirectional model explains several observations that remain puzzling under the sequential model alone. CGRP, which does not cross the blood–brain barrier, induces aura in a substantial proportion of participants with migraine with aura. The absence of aura induction in participants with migraine without aura and healthy volunteers further supports a threshold-based mechanism in which peripheral input must converge on a cortex already predisposed to CSD. 44
These findings indicate that peripheral activation is sufficient to precipitate CSD when cortical susceptibility is high, supporting a trigeminovascular → cortical direction of causality. The model also accommodates the preventive effects of anti-CGRP monoclonal antibodies, because blocking peripheral CGRP signaling reduces the likelihood of trigeminovascular activation and therefore reduces the chance of triggering CSD via thalamocortical modulation.51,52
Importantly, the bidirectional model does not negate the well-established ability of CSD to activate trigeminovascular pathways. Instead, it integrates both directions into a unified framework in which cortical and peripheral processes can each initiate attacks and amplify one another. CSD can activate meningeal nociceptors, and peripheral nociceptive activation can, under some conditions, facilitate CSD. This bidirectionality might explain the complex temporal relationships between aura and headache seen clinically, including aura during headache, aura without headache, and headache without aura. 53
Taken together, the available evidence positions the bidirectional neurovascular model as the most coherent framework for both provoked and spontaneous aura. It incorporates extensive preclinical data showing that CSD activates meningeal nociceptors. It also explains CGRP-induced aura through meningeal effects that can modulate thalamocortical excitability. This model further resolves the paradox created by the preventive effects of peripherally acting anti-CGRP monoclonal antibodies, which reduce aura despite minimal CNS penetration. Lastly, it offers a biologically plausible account of the inter-individual variability in aura susceptibility observed across provocation studies.
Strengths and limitations
This systematic review has several strengths. It provides the most comprehensive synthesis of human pharmacological provocation studies evaluating migraine aura under controlled lab conditions. By integrating findings across seven mechanistically distinct molecular triggers, the review offers a cohesive overview of how different molecular mechanisms might influence cortical susceptibility to aura.
Several limitations warrant consideration. First, the included studies were small, somewhat heterogeneous, and often open-label, increasing susceptibility to expectancy effects. Second, aura characterization was sometimes incomplete, with inconsistent reporting of symptom evolution, anatomical distribution, and duration. Third, provocation models might not fully recapitulate spontaneous attack dynamics, and mechanistic interpretations must therefore remain cautious. Finally, aura induction rates could depend on dose, timing, observation window, outcome definitions, and statistical power. Hence, negative provocation findings should not be interpreted as evidence against biological involvement of a pathway, particularly for agents studied in small or methodologically limited trials.
Conclusions
Human provocation studies show that migraine aura can be experimentally induced, with somewhat consistent responses arising from activation of CGRP signaling and vascular KATP channels. These potential aura triggers identify plausible mechanistic routes through which peripheral (meningeal) events might facilitate CSD. Larger studies are now needed to clarify these mechanisms, strengthen causal inference, and guide future research directions.
Article highlights
Human pharmacological provocation studies show that migraine aura can be experimentally induced, but far less frequently than migraine headache, highlighting distinct underlying mechanisms. Among tested agents, calcitonin gene-related peptide (CGRP) and pharmacological opening of vascular ATP-sensitive potassium (KATP) channels were seemingly able to induce migraine aura, implicating these pathways as the most plausible triggers. The findings support a bidirectional neurovascular model, in which meningeal trigeminovascular signaling might lower cortical spreading depolarization thresholds in people with migraine with aura, refining current concepts of migraine aura pathogenesis.
Supplemental Material
sj-docx-1-cep-10.1177_03331024261429112 - Supplemental material for Molecular triggers of migraine aura: A systematic review of human pharmacological provocation studies
Supplemental material, sj-docx-1-cep-10.1177_03331024261429112 for Molecular triggers of migraine aura: A systematic review of human pharmacological provocation studies by Zahra Hakimzadeh, Basit Ali Chaudhry, Haidar M. Al-Khazali, Rune Häckert Christensen, Messoud Ashina and Håkan Ashina in Cephalalgia
Footnotes
Abbreviations
Acknowledgements
Not applicable.
Author contributions
ZH: Data curation; Investigation; Methodology; Validation; Writing – original draft; Writing – review & editing.
BAC: Data curation; Investigation; Validation; Writing – review & editing.
HMA: Methodology; Supervision; Writing – review & editing.
RHC: Methodology; Writing – review & editing.
MA: Conceptualization; Funding acquisition; Methodology; Supervision; Writing – review & editing.
HA: Conceptualization; Funding acquisition; Methodology; Project administration; Supervision; Writing – review & editing.
All authors meet ICMJE authorship criteria and approved the final manuscript.
Consent to participate
Not applicable.
Consent for publishing
All authors have read and approved the final version of this manuscript and consent to its submission and publication. The corresponding author accepts responsibility for ensuring that all authors provide informed consent for publication.
Data availability statement
Upon reasonable request, the corresponding author will provide the necessary data and materials to interested researchers for the purpose of academic scrutiny, reproducibility, and further scientific investigation.
Declaration of conflicting interests
BAC has received personal fees from Pfizer, outside the submitted work. HMA has received personal speaker fees from AbbVie, Pfizer, and Lundbeck, outside of the submitted work. RHC has received personal speaker fees from AbbVie, Pfizer, Lundbeck, and Teva, outside of the submitted work. MA has also received institutional research grants from the Danish National Research Foundation, Novartis, Novo Nordisk Foundation, and Lundbeck Foundation. MA is also an Associate Editor of Brain and The Journal of Headache and Pain. HA has received personal fees from AbbVie, Lundbeck, Pfizer and Teva, outside of the submitted work. HA is also an Editorial Board Member of The Journal of Headache and Pain. The remaining authors declare no conflicts of interest.
Ethical considerations
Not applicable for reviews.
Funding
Funding was provided by the Danish National Research Foundation (DNRF188 to MA) and Lundbeck Foundation (R310-2018-3711 to MA; R403-2022-1352 and R481-2024-1392 to HA). The funding sources were not involved in the design or conduct of the study; in the collection, analysis, or interpretation of the data; or in the preparation, review, or approval of the manuscript.
Open practices
Not applicable.
ORCID iDs
Supplemental material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.