
Abstract
Background
Cortical spreading depolarization, the cause of migraine aura, is a short-lasting depolarization wave that moves across the brain cortex, transiently suppressing neuronal activity. Prophylactic treatments for migraine, such as topiramate or valproate, reduce the number of cortical spreading depression events in rodents.
Objective
To investigate whether cortical spreading depolarization with and without chronic treatment with topiramate or valproate affect the DNA methylation of the cortex.
Methods
Sprague-Dawley rats were intraperitoneally injected with saline, topiramate or valproate for four weeks when cortical spreading depolarization were induced and genome-wide DNA methylation was performed in the cortex of six rats per group.
Results
The DNA methylation profile of the cortex was significantly modified after cortical spreading depolarization, with and without topiramate or valproate. Interestingly, topiramate reduced by almost 50% the number of differentially methylated regions, whereas valproate increased them by 17%, when comparing to the non-treated group after cortical spreading depolarization induction. The majority of the differentially methylated regions lay within intragenic regions, and the analyses of functional group over-representation retrieved several enriched functions, including functions related to protein processing in the cortical spreading depolarization without treatment group; functions related to metabolic processes in the cortical spreading depolarization with topiramate group; and functions related to synapse and ErbB, MAPK or retrograde endocannabinoid signaling in the cortical spreading depolarization with valproate group.
Conclusions
Our results may provide insights into the underlying physiological mechanisms of migraine with aura and emphasize the role of epigenetics in migraine susceptibility.
Keywords
Introduction
Migraine is a common disabling neurological disorder characterized by recurrent attacks of severe unilateral headache that are preceded by premonitory symptoms. Approximately 30% of migraine sufferers experience transient neurological disturbances manifested as visual, sensory or motor symptoms, that usually occur before the headache: the migraine aura (1). Cortical spreading depolarization (CSD), considered the cause of migraine aura (2), is a short-lasting neuronal and glial depolarization wave that moves across the brain cortex which is followed by a wave of depression of evoked and spontaneous electroencephalogram activity (3). CSD has been long hypothesized to be the trigger of headache mechanisms (4), especially due to its persistent activation of meningeal nociceptors (5); although this is still debated especially because most migraine sufferers do not experience aura and it is unlikely that CSD is involved in initiating the complete syndrome of migraine (6).
Several studies have demonstrated the existence of genetic factors that contribute to the susceptibility to migraine (7). Over the last years, genome-wide association studies (GWAS) in large migraine case-control cohorts have increased our understanding of the variety of genetic factors that may contribute to common migraine (8), but there still remains uncertainty about the causal genes involved. Besides this genetic load, migraine susceptibility might also be modulated by internal and external non-genetic factors, such as sex hormones, lifestyle and environmental changes (9–11), which could have an impact through epigenetic mechanisms.
DNA methylation involves the transfer of a methyl group onto cytosine to form 5-methylcytosine, which in turn regulates gene expression by inhibiting the binding of transcription factors to DNA and by recruiting proteins involved in gene repression (12). Changes in DNA methylation have been linked to a wide variety of diseases, including neurological diseases such as stroke and epilepsy (13,14). Recently, genome-wide DNA methylation studies in blood samples were used to analyze potential differences between migraine sufferers and controls (15) and potential changes associated with headache chronification (16,17), without finding any significant loci associated. The main limitation of these studies is the use of blood samples, due to the obvious difficulty in obtaining the target tissue, and that the concordance of the epigenetic state between the brain tissue and blood is still not clear (18). Hence, the use of a rodent model of migraine is a useful approach to study the epigenetic signatures in the brain. In support of this, a study has shown that the methylation status of migraine-related genes in rat, including Crcp, Calcrl, Esr1 and Nos3, is tissue-specific and that the methylation in rat leukocytes is similar to that in humans (19).
Several studies have proven that preventive antimigraine drugs, including the anti-epileptics valproate (VPA) and topiramate (TPM), suppress CSD when administered chronically (for four weeks), but not acutely, in preclinical models of rat and cat (20–24). This need for chronic administration prompted us to hypothesize that the mechanisms involved in migraine prevention might be acting at a genomic level, involving epigenetic mechanisms such as modifying the DNA methylation status of genes that could be related to migraine susceptibility.
With the aim to understand whether epigenetic changes may be related to migraine aura with or without the treatment with antimigraine prophylactics, we explored the short-term effects of cortical spreading depolarization (one hour after the induction) in DNA methylation in the cortex of saline, valproate and topiramate treated animals.
Materials and methods
Experimental groups
Thirty-six adult male Sprague-Dawley rats (Charles River Laboratories, Spain) received daily intraperitoneal injections for four weeks (chronic treatment) of saline (Fisiológico Braun 0.9%, B Braun, Spain), valproate (VPA, 200 mg/kg; Sanofi-aventis, Spain) or topiramate (TPM, 80 mg/kg; gift by Janssen-Cilag, Spain); chosen dosages were based on previous literature (21). Rats were randomly divided into four experimental groups:
saline-SHAM: injected with saline for four weeks, underwent surgery without CSD-induction saline + CSD: injected with saline for four weeks, underwent surgery with CSD-induction VPA + CSD: injected with valproate for four weeks, underwent surgery with CSD-induction TPM + CSD: injected with topiramate for four weeks, underwent surgery with CSD-induction
Animals were housed in a temperature- and humidity-controlled facility with a 12 h light cycle and with ad libitum access to food and water. All experimental procedures were approved by the Vall d’Hebron Institute of Research Animal Experimentation Ethics Committee and guidelines for animal care were strictly followed.
Anesthesia and surgery
On the day of the experiment, rats were anesthetized using isoflurane (4.5% induction, 1–1.5% maintenance, in 70% N2O/30% O2) and the depth of anesthesia was monitored throughout the whole experiment by checking the hind paw and blink reflexes. Temperature was kept at 37°C using a heating pad. Rats were placed in a stereotaxic frame (David Kopf Instruments, USA) and two burr holes were drilled over the right hemisphere at the following coordinates (from bregma):
posterior 5 mm, lateral 2 mm: KCl application site posterior 0.5 mm, lateral 2 mm: recording site
Dura overlying the cortex was gently removed and care was taken to avoid bleeding.
Electrophysiological recordings
Cortical direct current (DC) potential shifts were recorded as previously described (25). Briefly, glass micropipettes (Harvard apparatus, USA) filled with NaCl 4M were placed 500 µm below the dural surface for DC recordings. An Ag/AgCl reference electrode (World Precision Instruments, USA) was placed subcutaneously in the neck. After surgical preparation, the cortex was allowed to recover for 15 minutes under saline irrigation before the start of the recording. The data obtained were amplified, digitized and recorded using a data acquisition system (Neurolog system, UK) and were displayed on an online data analysis system (CED Spike2 v7.03 software).
CSD induction
CSDs were induced by placing a cotton ball soaked with KCl 1M over the pial surface at the application site, which was kept moist adding an extra 5 µl every 15 minutes. The total number of KCl-induced CSDs was counted during one hour of KCl application and comparisons were performed between groups.
Samples
After one hour of recordings, animals were euthanized by decapitation and brains were gently removed from the skull. The right cortices that underwent CSD-induction were dissected (without including the small region where the KCl was applied), flash frozen in liquid nitrogen and mechanically homogenized in liquid nitrogen with a mortar and pestle. DNA was extracted from the tissue powder by using the QIAamp DNA mini kit (Qiagen, Spain) according to the manufacturer’s specifications. Concentration and integrity of DNA was determined on a Fluostar Optima plate reader (BMG Labtech, Germany) with the Quant-iT™ Picogreen dsDNA assay kit (Invitrogen, Belgium) on 480/520 nm.
Genome-wide Methyl-CpG-binding domain sequencing (MBD-sequencing)
Six rats were randomly selected from each experimental group to carry out Methyl-CpG-binding domain sequencing (MBD-sequencing). 3 µg of DNA were fragmented on Covaris S2 (Covaris, USA) with the following settings: duty cycle 10%, intensity 5, 200 cycles per burst for 190 seconds, to obtain fragments with an average length of 200 bp. Then, methylated-enriched DNA was captured using the MethylCap kit (Diagenode, Belgium) and 250 ng were used to perform library preparation on Apollo 324 with PrepX-DNA Library Kit (IntegenX, USA). Library amplification was performed according to multiplexed paired end ChIP protocol. Samples were adjusted to 10 nM and sequenced in an Illumina Hi-Seq 2000 (Illumina, USA).
Mapping and peak calling for MBD-sequencing enrichment
FASTQ sequence reads were generated using the IlluminaCasava pipeline version 1.8.0. Paired end 51 bp sequence reads were mapped to the rat genome (UCSC assembly rn4, NCBI build RGSC_v3.4) using Bowtie v0.12.9 software. Bowtie files were converted into bam format.
MBD-sequencing quality controls: Saturation analysis and CpG enrichment
Three different approaches were used to perform quality controls of MBD-sequencing by using the MEDIPS R software v1.10.0 (26). First, a saturation analysis was used to determine if the input sequence data coverage was sufficiently deep for reproducibility. Second, the CpG coverage rate was calculated by testing the number of CpGs covered by the given reads and the depth of coverage per CpG. Finally, the CpG enrichment was calculated to ensure that the assays enriched the samples for CpG sites as expected. Quality control parameters were considered correct if saturation >0.5, 5x CpG coverage rate >0.05 and CpG enrichment >1.4. Threshold cut-off values were based on previous literature (27).
Identification and annotation of differentially methylated regions (DMRs)
To identify differentially methylated regions (DMRs) between groups, the bam files obtained from MBD-sequencing were analyzed with the MEDIPS R v1.10.0 software. DMRs were considered significant when the false discovery rate (FDR) ≤ 0.05, the fold change ≥2 and regions were methylated at least in four out of the six rats of the same group. Genomic feature data was retrieved from UCSC Genome Browser database (assembly rn4) to annotate the genes described, by using RefSeq Genes, Ensembl Genes, RGD Genes, tRNA Genes and miRNA tracks; the genes predicted, by using Genscan, Geneid Genes and SGP Genes tracks; CpG islands (CGIs), by using CpG Islands and CpG Islands (AL) tracks; the proximal promoters, which were defined as regions within 1 kb upstream and 500 pb downstream from the transcription start site of described genes; and the distal promoters, which were defined as regions within 5 kb upstream and 500 pb downstream from the transcription start site of described genes; with MEDIPS R software.
Hierarchical clustering and principal component analysis
Heatmap representation of the DMRs in the different comparisons was performed using the package gplots in R/Bioconductor. Average linkage clustering and Manhattan distance measure was applied and color display was calculated per relative comparison in each row (per DMR). Principal component analysis was made using the built-in R/Bioconductor function prcomp() (variables were not scaled before the analysis).
Pathway analysis
All genes described containing DMRs within their intronic or exonic regions were analyzed for functional enrichment analysis in Gene Ontology and KEGG Reactome by using the g:Profiler web server (28). This program calculates a minimum hypergeometric p value for each pathway, placing greater weight on top-ranked genes where enrichment is the strongest, allowing the detection of small and highly significant pathways and larger pathways that are more representative of the entire gene list (15). We set the minimum overlap between our list of genes and Gene Ontology and KEGG Reactome pathways as n = 2 with false discovery rate ≤ 0.05 to adjust for multiple testing.
Bisulfite pyrosequencing (BPS) validation
To validate the DNA methylation data obtained from MBD-Sequencing, bisulfite pyrosequencing (BPS) analysis was performed bisulfite-converting genomic DNA using EpiTectPlus Bisulfite Conversion Kit (Qiagen, Germany) according to manufacturer’s specifications. A subsequent PCR amplification was performed using biotinylated primers (primer sequences are shown in online Supplementary Table 1). Pyrosequencing and data analysis were performed with the pyrosequencer analyzer PyroMark Q96 (Qiagen, Germany) according to manufacturer’s instructions.
DMRs within genes Itgav, Kcnd2, Pcdhb3 and Rimbp2, which were differentially methylated in the saline + CSD vs VPA + CSD comparison, and within genes Anks1b and Ndufs3, which were differentially methylated in the saline + CSD vs TPM + CSD comparison, were validated using six rat brain samples per comparison.
Statistical analysis
Statistical analysis of data was performed using IBM SPSS 22.0 and R software 3.0.1 and graphs were plotted with SigmaPlot 12.5 and R software. For each study, N was chosen to obtain a statistical power of 80-90% and analyses of power were performed with G*Power software.
For CSD experiments, the number of KCl-induced CSDs were counted over one hour provided that the cortical steady state potential was altered by at least 10 mV.
For group comparisons (CSD experiments, overall methylation levels and BPS validation) normality was tested using the Kolmogorov-Smirnov test. If data was normally distributed, an independent Student’s t test or an ANOVA for multiple comparisons were used (data expressed as mean ± SEM). If data was not normally distributed, we used Mann Whitney U test for comparison with Bonferroni post-hoc correction (data expressed as a median with interquartile range). P-value was considered significant if ≤0.05.
DMRs were considered significant when adjusted p value for multiple comparisons ≤0.05, false discovery rate (FDR) ≤0.05, fold change ≥2 and regions were methylated at least in four out of the six replicates.
Results
Valproate and topiramate reduced CSD frequency
Daily injections of valproate and topiramate for four weeks significantly reduced the CSD frequency in rats (Figure 1), as previously described (21). Specifically, topiramate reduced the number of CSDs from 12.8 ± 1.7 to 9 ± 1.3 (compared to saline + CSD group, t(10) = 4.4, P = 0.001) and valproate from 12.8 ± 1.7 to 6.67 ± 2.2 (compared to saline + CSD group, t(10) = 5.5, P = 0.0002).
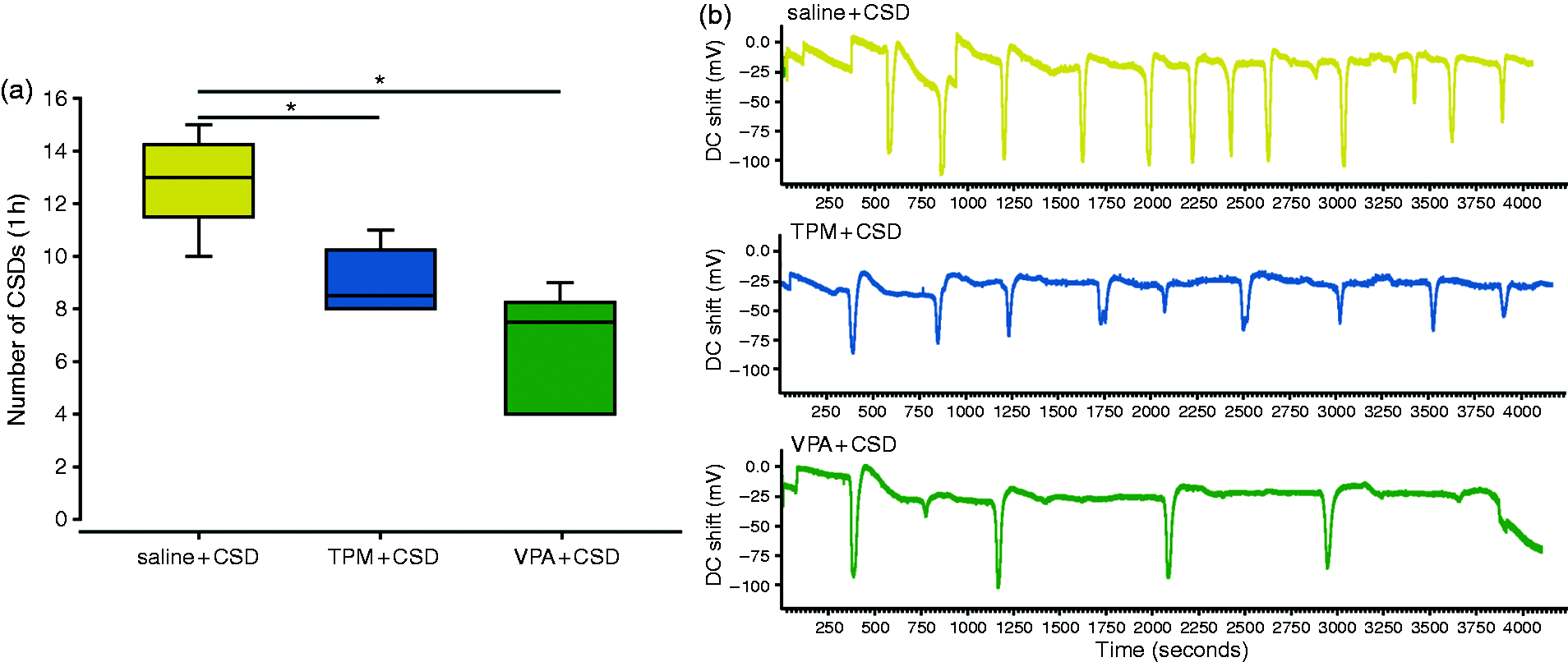
Valproate and topiramate reduced the KCl-induced CSD frequency. (a) The number of CSDs occurring during one hour of KCl induction was significantly reduced after the four-week pre-treatment with topiramate (TPM + CSD) and valproate (VPA + CSD) when compared to the saline + CSD group (saline + CSD). (b) In vivo cortical direct current (DC) shift readings in a saline + CSD, a TPM + CSD and a VPA + CSD representative animal showing application of a cotton ball soaked in KCl on the cortex surface inducing multiple CSDs throughout the one-hour recording. (Data are expressed as mean ± SEM; *: P < 0.05, TPM: topiramate, VPA: valproate).
Genome-wide DNA methylation analysis
Genome-wide DNA methylation profiles of all right cerebral cortices were determined by using Methyl-CpG-binding domain sequencing (MBD-sequencing). All samples correctly passed the quality control analyses, total and mapped reads are available in online Supplementary Table 2.
Taking into account all the regions sequenced and mapped, there were no significant differences in the overall methylation levels between groups (F3,23 = 1.13, P = 0.36). The genomic distribution of the methylated regions was analyzed and classified between intergenic, intronic, exonic and promoter regions. The highest percentage of methylated regions lay within intergenic regions (58%), followed by introns (31.9%), exons (7.1%) and promoters (2.9%), without significant differences between groups (intergenic regions: F3,23 = 2.6, P = 0.08, introns: F3,23 = 0.336, P = 0.8, exons: F3,23 =1.41, P = 0.27, promoters: F3,23 = 1.05, P = 0.391) (Figure 2a), which is consistent with previously published genome-wide methylation studies (29).

Unsupervised analysis of the methylated regions sequenced with MBD-Sequencing. (a) The methylated regions were classified according to their genomic distribution, with most of them lying within intergenic regions, followed by intronic, exonic and promoter regions, without finding significant differences between the four groups. (b) Results of the principal components 1 and 2 of the Principal Component Analysis (PCA) of the methylated regions of all the samples included in the study.
We analyzed all the sequenced regions of the four groups using an unsupervised principal component analysis (PCA), which did not classify the samples according to their groups (Figure 2b).
Early effects of CSD on DNA methylation
With the aim to detect the early effects of CSD on the DNA methylation of the cerebral cortex, we compared the genome-wide DNA methylation profiles of the saline + CSD group with the saline-SHAM group (control). Interestingly, we found 283 differentially methylated regions (DMRs) between both groups. This high number of DMRs suggests that CSD induces a significant and fast modulation of DNA methylation in cortical cells.
Firstly, we performed a supervised hierarchical clustering of the DMRs that revealed significant methylation changes between the saline + CSD and the saline-SHAM groups and that correctly classified the six samples within their group (Figure 3a). We then proceeded to characterize the DMRs found in this comparison (Figure 3b–e). Interestingly, of the total 283 DMRs, 70% were hyper-methylated in the saline + CSD group and a big majority of them (82%) were classified as intragenic (Figure 3b–c). From the intragenic DMRs, 37% where found within described genes (Figure 3d) of which 21 were found hypomethylated and 22 hypermethylated (Figure 3e). The complete list of DMRs with their associated genomic features is listed in online Supplementary Table 3.
The analysis of the functional group over-representation retrieved several enriched functions that are mainly linked to protein processing, including “post-translational protein modification” and “protein localization to synapse”, among others (Figure 3f).
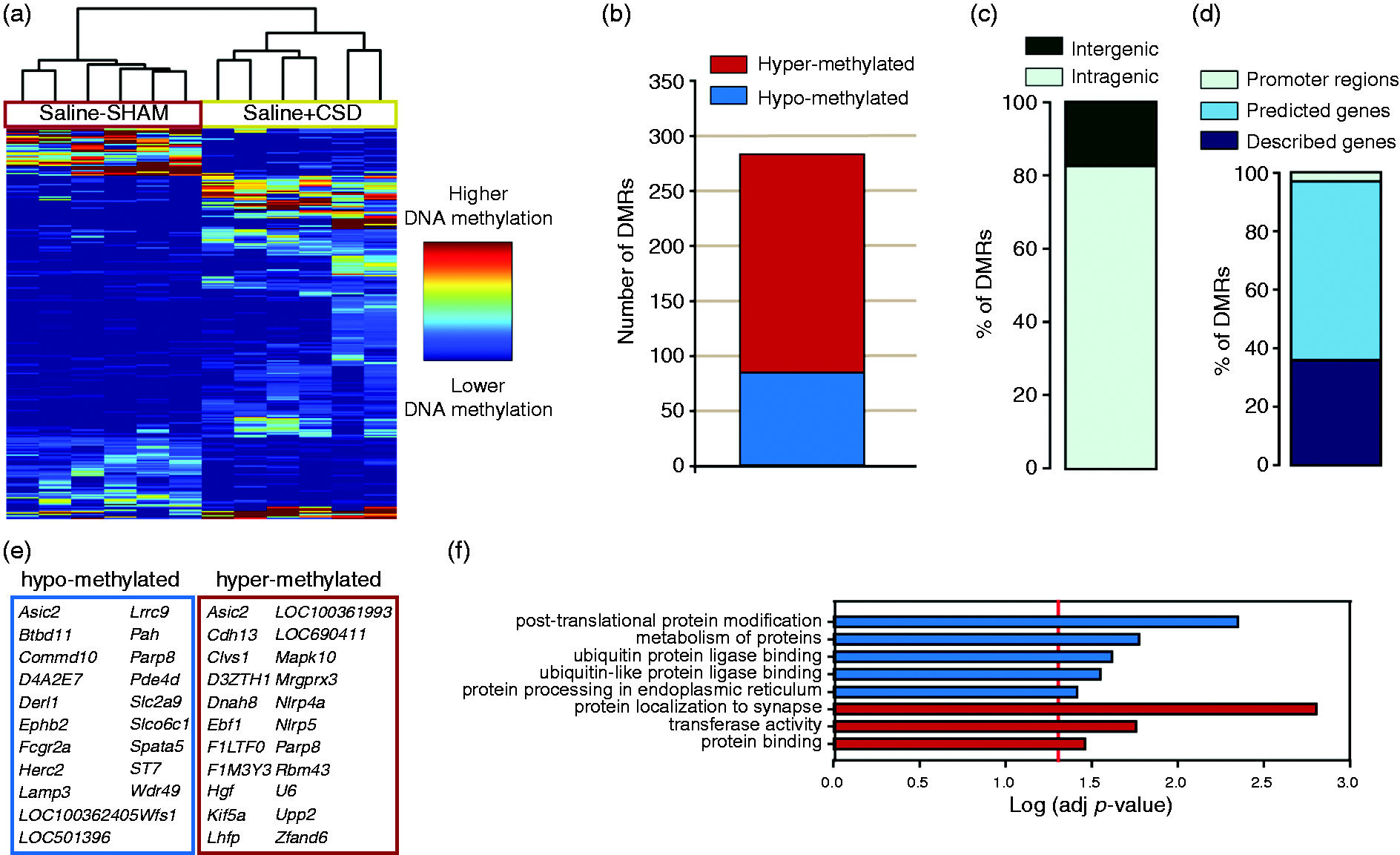
Early effects of CSD on DNA methylation: results of the comparison of the genome-wide DNA methylation profiles of the saline+CSD with the saline-SHAM groups. (a) Supervised hierarchical clustering of the DMRs according to their relative methylation score. (b) Number of DMRs hyper- or hypo-methylated within the saline + CSD group. (c–d) Classification of the DMRs according to their genomic localization. (e) List of genes with DMRs. (f) List of functions retrieved by the functional group over-representation with the Log (adjusted P-value) obtained in the analysis.
Modulation of the early effects of CSD on DNA methylation induced by chronic topiramate treatment
We then analyzed if the chronic pre-treatment with topiramate could modulate the changes in DNA methylation induced by CSD. To study this, we compared the genome-wide DNA methylation profiles of the TPM + CSD with the saline + CSD group. In this comparison, we found 151 DMRs between both groups, indicating a 47% decrease in the number of DMRs when compared to the saline + CSD vs saline + CSD comparison.
The supervised hierarchical clustering of the DMRs correctly classified the six samples within their group, revealing significant methylation changes between both groups (Figure 4a). Opposite to the ratio found in the previous comparison, of the total 151 DMRs, only 30% were hyper-methylated in the TPM + CSD group (Figure 4b). However, the same percentage of DMRs was found within intragenic (82%) regions (Figure 4c), with also a similar percentage lying in described genes (Figure 4d). In this group, there were 19 genes with hypomethylated regions and seven with hypermethylated regions (Figure 4e). The complete list of DMRs with their associated genomic features is listed in online Supplementary Table 4.

Modulation of the early effects of CSD on DNA methylation induced by chronic TPM treatment: results of the comparison of the genome-wide DNA methylation profiles of the TPM + CSD with saline + CSD the groups. (a) Supervised hierarchical clustering of the DMRs according to their relative methylation score. (b) Number of DMRs hyper- or hypo-methylated within the TPM + CSD group. (c-d) Classification of the DMRs according to their genomic localization. (e) List of genes with DMRs. (f) List of functions retrieved by the functional group over-representation with the Log (adjusted P-value) obtained in the analysis. (g) The validation using bisulfite pyrosequencing showed significant differential methylation levels within the same direction as those found with MBD-Sequencing (Data expressed as mean ± SEM; *: P < 0.05).
The analysis of the functional group over-representation retrieved functions that are mainly linked to several metabolic processes, to Rap1 signaling pathway and to the regulation of response to stimulus, among others (Figure 4f).
We selected two DMRs from MBD-sequencing to be validated with bisulfite pyrosequencing. All the regions that were analyzed were correctly validated, showing differential methylation levels within the same direction as found with MBD-sequencing (Figure 4g).
Modulation of the early effects of CSD on DNA methylation induced by chronic valproate treatment
We then analyzed if the chronic pre-treatment with valproate could modulate the changes in DNA methylation induced by CSD, as we had seen with topiramate. To study this, we compared the genome-wide DNA methylation profiles of the VPA + CSD with the saline + CSD group. In this comparison, we found 332 DMRs between both groups, indicating a 17% increase in the number of DMRs when compared to the saline + CSD vs saline-SHAM comparison. The initial supervised hierarchical clustering of the DMRs also revealed significant methylation changes between the VPA + CSD and the saline + CSD group, correctly classifying the six samples within their group (Figure 5a).

Modulation of the early effects of CSD on DNA methylation induced by chronic VPA treatment: results of the comparison of the genome-wide DNA methylation profiles of the VPA + CSD with the saline + CSD groups. (a) Supervised hierarchical clustering of the DMRs according to their relative methylation score. (b) Number of DMRs hyper- or hypo-methylated within the VPA + CSD group. (c-d) Classification of the DMRs according to their genomic localization. (e) List of genes with DMRs. (f) List of functions retrieved by the functional group over-representation with the Log (adjusted P-value) obtained in the analysis. (g) The validation using bisulfite pyrosequencing showed significant differential methylation levels within the same direction as those found with MBD-Sequencing (Data expressed as mean ± SEM; *: P < 0.05).
Similarly to the initial comparison (saline + CSD vs saline-SHAM), of the total 332 DMRs, 62% were hyper-methylated in the VPA + CSD group (Figure 5b). Also, of the total DMRs, 83% were intragenic (Figure 5c) and of those 32% were found within described genes (Figure 5d). Of these genes, 13 had hypo-methylated regions and 33 hyper-methylated regions (Figure 5e). The complete list of DMRs with their associated genomic features is listed in online Supplementary Table 5.
The analysis of the functional group over-representation retrieved several functions, including “ErbB signaling pathway”, “synapse part”, “MAPK signaling pathway” and “retrograde endocannabinoid signaling”, among others (Figure 5f).
We selected two DMRs from MBD-sequencing to be validated with bisulfite pyrosequencing. All the regions that were analyzed were correctly validated, showing differential methylation levels within the same direction as found with MBD-sequencing (Figure 5g).
Discussion
In this study, we investigated the epigenetic effects of cortical spreading depolarization (CSD), as a surrogate for migraine with aura, on genome-wide DNA methylation in the cerebral cortex of a well-validated preclinical model of migraine. To our knowledge, this is the first study to assess CSD short-term effects in genome-wide DNA methylation with and without the prophylactic treatments topiramate (TPM) and valproate (VPA).
CSD has been long hypothesized to modulate different epigenetic mechanisms. Our results are in line with this hypothesis and show that CSD has a significant and fast effect on the modulation of DNA methylation in the cerebral cortex where the CSDs were induced, which can be modified with the use of the anti-migraine prophylactic treatments TPM and VPA. At the transcriptomic level, several studies have confirmed that CSD modulates gene expression in the cerebral cortex of rodents. Specifically, CSD has been shown to modulate the expression of genes involved in inflammatory processes (30), in sleep homeostasis (31), in neuroprotection (32) and in stress responses (33), amongst others. In agreement with this, our group showed in a previous study that CSD induction in the same samples used in this study modulates the expression of genes related to hormone stimulus, apoptosis, synaptic transmission and interleukin signaling, without differences between the animals pre-treated or not with TPM or VPA (34).
CSD could be affecting gene expression by modulating several mechanisms, including epigenetic mechanisms. A previous study confirmed that histone methylation is one of these epigenetic mechanisms that are modulated by CSD, specifically the methylation of histones H3K4 and H3K9, which are directly correlated with changes in gene transcription rates (35). Our results confirm that DNA methylation is also modulated by CSD as they show that the induction of multiple CSDs produce significant short-term effects in genome-wide DNA methylation. Specifically, CSDs have shown to alter the methylation pattern of 283 genomic regions in comparison with animals without undergoing CSDs, with 30% of them being hyper-methylated in the saline-SHAM group. Almost 80% of these differentially methylated regions (DMRs) were classified as intragenic, although only a low proportion lay within described genes, which is in line with previous genome-wide DNA methylation studies (29). Regarding the described genes with DMRs, the list includes ion channels expressed in the CNS (Asic2); genes involved in nociception (Mrgprx3p); and genes involved in neural plasticity (Ephb2, Cdh13 and Mapk10); among others (see the whole list in Figure 3e). Regarding the functional group over-representation analysis of genes containing DMRs, most of the functions that were retrieved correspond to the post-translational modification and processing of proteins, without finding any relevant term that could be directly involved in CSD or migraine pathophysiology.
Our analysis also shows that the pre-treatment with TPM for four weeks, besides the expected significant reduction of the number of CSDs, induced a 47% decrease in the number of DMRs when compared to the saline-SHAM vs saline + CSD comparison, with 30% of them being hyper-methylated in the TPM + CSD group compared to saline + CSD group. This is an interesting finding, showing for the first time the role of TPM on brain DNA methylation, as to date TPM had only been characterized as a histone deacetylase (HDAC) inhibitor (36). As in the previous comparison, most of the DMRs were found within intragenic regions with a low proportion lying within described genes. The list of genes with DMRs includes genes involved in signaling cascades (Fgfr2, Grip1, Phlpp1 and Ppm1l); in transcriptional regulation (Pbx3, Scmh1 and Smbt1); in the regulation of homeostasis during prolonged fasting (Sirt5); and in the regulation of dendritic spine morphogenesis (Sipa1l1); among others (see the whole list in Figure 4e). Regarding the functional group over-representation analysis of genes with DMRs, most of the functions that were retrieved are mainly linked to several metabolic processes, to Rap1 signaling pathway and to the regulation of response to stimulus, again without finding any term that could be directly involved in CSD or migraine pathophysiology.
Our results also show that the pre-treatment with VPA for four weeks, besides the expected significant reduction of the number of CSDs, induced a 17% increase in the number of DMRs when compared to the saline-SHAM vs saline + CSD comparison, with 62% of them being hyper-methylated in the VPA + CSD group compared to the saline + CSD group. Although none of the significant DMRs found in the saline + CSD vs VPA + CSD comparison were the same that those found in the saline-SHAM vs saline + CSD comparison, the genomic profile of the DMRs was very similar, with 76% of the DMRs classified as intragenic and also only a low proportion lying within described genes. Of particular interest, the list of genes with DMRs include Ephb2, which has been previously shown to contribute to the synaptic plasticity of chronic migraine through NR2B phosphorylation (37); Piezo2, which is from the family of Piezo channels that have been proposed to underlie the mechanotransduction in migraine pathophysiology (38); and Prkca, which modulates the activity of ASIC1 and ASIC3, the acid-sensing ion channels that have been linked to migraine pathophysiology (39).The list of genes with DMRs also includes Gabrg1, which encodes the receptor for the neurotransmitter GABA, and genes involved in RNA-mediated gene silencing (Ago3); in inflammation (Faslg); in the regulation of transcription (Med15); in the regulation of the circadian clock (Relb); in axon guidance (Spon1); and in synaptogenesis (Syn3) (see the whole list in Figure 5e). Regarding the functional group over-representation analysis of genes with DMRs, the only term that could be related to migraine pathophysiology is “retrograde endocannabinoid signaling”, as the endocannabinoid system has been found to interact with the serotonergic system in the processing of somatosensory nociceptive information (40).
In this study, differences in methylation due to the effect of prophylactic treatments were not investigated. Hence, our experimental design allowed us to identify differences after treatment and CSD, but not to distinguish which alterations are induced by the treatment, nor due to the CSD events. However, our results show that in the group pre-treated with TPM there is a reduction in the number of DMRs and in the group pre-treated with VPA there is an increase in the number of DMRs, even when in both cases there was a significant reduction in the number of CSDs. Although these results may point towards an effect of the treatment on the reduction/increase of the number of DMRs, to definitively confirm this, future experiments including extra groups of animals with chronic treatment of TPM or VPA without CSD induction should be analyzed. Our results should be interpreted in the context of several methodological considerations. Some limitations include: 1) the time point after the CSD induction, one hour, means our results show the short-term effects of CSD, with no focus on long-term consequences; 2) the cortex samples included the whole right cortex in which CSD was induced, without narrowing on specific areas of it as a previous study had done (33); 3) the effects of the treatment before the CSD were not assessed, hence the effects of the treatment itself could not be analyzed; 4) we assessed the effects of multiple CSDs which may not be comparable with spontaneous CSD in migraine patients; 5) animals were anesthetized with isoflurane during CSD induction, which might have affected their susceptibility to CSD-induction; 6) the number of animals used per group is limited (N = 6), although similar and higher to previous studies (41–44). However, several strengths of our study reinforce the validity of our results, including: 1) the results identified overcome highly restrictive multiple-testing corrections and substantial fold-change threshold (>2); 2) validation of two DMRs by another technique confirmed the results obtained by MBD-seq; 3) our results are consistent with previous studies assessing CSD-induced transcriptomic modifications, including short-term modifications (33,34).
Conclusions
Our study shows that CSD induces short-term effects on DNA methylation in the cerebral cortex of rats, consistent with previous literature showing effects of CSD on epigenetic modifications, that can be modified by pre-treating the animals with prophylactic drugs (TPM or VPA), emphasizing the role of epigenetics in migraine susceptibility. Further studies assessing different time points may help to better understand the course of DNA methylation changes that take place when CSD is triggered with or without prophylactic treatment. We consider that assessing the changes induced by CSD with or without prophylactic treatments in a preclinical model of migraine aura may provide an insight on the underlying physiological mechanisms of this disorder, although the translation of our findings would require further investigation.
Article highlights
CSD induces short-term effects on DNA methylation in the cerebral cortex of rats. Chronic treatment with topiramate or valproate modify the effects of CSD-induced changes in DNA methylation.
Supplemental Material
sj-xlsx-1-cep-10.1177_03331024221146317 - Supplemental material for Genome-wide DNA methylation analysis in an antimigraine-treated preclinical model of cortical spreading depolarization
Supplemental material, sj-xlsx-1-cep-10.1177_03331024221146317 for Genome-wide DNA methylation analysis in an antimigraine-treated preclinical model of cortical spreading depolarization by Marta Vila-Pueyo, Ester Cuenca-León, Ana C. Queirós, Marta Kulis, Cèlia Sintas, Bru Cormand, José Ignacio Martín-Subero, Patricia Pozo-Rosich, Noèlia Fernàndez-Castillo, Alfons Macaya in Cephalalgia
Supplemental Material
sj-xlsx-2-cep-10.1177_03331024221146317 - Supplemental material for Genome-wide DNA methylation analysis in an antimigraine-treated preclinical model of cortical spreading depolarization
Supplemental material, sj-xlsx-2-cep-10.1177_03331024221146317 for Genome-wide DNA methylation analysis in an antimigraine-treated preclinical model of cortical spreading depolarization by Marta Vila-Pueyo, Ester Cuenca-León, Ana C. Queirós, Marta Kulis, Cèlia Sintas, Bru Cormand, José Ignacio Martín-Subero, Patricia Pozo-Rosich, Noèlia Fernàndez-Castillo, Alfons Macaya in Cephalalgia
Supplemental Material
sj-xlsx-3-cep-10.1177_03331024221146317 - Supplemental material for Genome-wide DNA methylation analysis in an antimigraine-treated preclinical model of cortical spreading depolarization
Supplemental material, sj-xlsx-3-cep-10.1177_03331024221146317 for Genome-wide DNA methylation analysis in an antimigraine-treated preclinical model of cortical spreading depolarization by Marta Vila-Pueyo, Ester Cuenca-León, Ana C. Queirós, Marta Kulis, Cèlia Sintas, Bru Cormand, José Ignacio Martín-Subero, Patricia Pozo-Rosich, Noèlia Fernàndez-Castillo, Alfons Macaya in Cephalalgia
Supplemental Material
sj-xlsx-4-cep-10.1177_03331024221146317 - Supplemental material for Genome-wide DNA methylation analysis in an antimigraine-treated preclinical model of cortical spreading depolarization
Supplemental material, sj-xlsx-4-cep-10.1177_03331024221146317 for Genome-wide DNA methylation analysis in an antimigraine-treated preclinical model of cortical spreading depolarization by Marta Vila-Pueyo, Ester Cuenca-León, Ana C. Queirós, Marta Kulis, Cèlia Sintas, Bru Cormand, José Ignacio Martín-Subero, Patricia Pozo-Rosich, Noèlia Fernàndez-Castillo, Alfons Macaya in Cephalalgia
Supplemental Material
sj-xlsx-5-cep-10.1177_03331024221146317 - Supplemental material for Genome-wide DNA methylation analysis in an antimigraine-treated preclinical model of cortical spreading depolarization
Supplemental material, sj-xlsx-5-cep-10.1177_03331024221146317 for Genome-wide DNA methylation analysis in an antimigraine-treated preclinical model of cortical spreading depolarization by Marta Vila-Pueyo, Ester Cuenca-León, Ana C. Queirós, Marta Kulis, Cèlia Sintas, Bru Cormand, José Ignacio Martín-Subero, Patricia Pozo-Rosich, Noèlia Fernàndez-Castillo, Alfons Macaya in Cephalalgia
Footnotes
Acknowledgements
The authors thank M Jiménez and D Gallego for providing the glass micropipettes, A Andreou for her technical support during the CSD recordings and X de Pedro and A Pérez-Pineda for their support during the statistical analysis.
Declaration of conflicting interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: The work was funded by the Spanish Ministry of Science and Innovation, the Spanish Ministry of Economy and Competitiveness, Fondos Europeos de Desarrollo Regional (FEDER) Funds, and Plan E (Grants SAF2009-13182-C01 and C03, SAF2012-31089, SAF2012-38140), Fondo de Investigación Sanitaria (Red HERACLES RD12/0042/0014) and Generalitat de Catalunya (grants 2009SGR1369, 2009SGR0971 and 2009SGR0078). MV-P was supported by a predoctoral grant from VHIR, Barcelona (Spain), and by the European Union’s Horizon 2020 research and innovation programme under the Marie Skłodowska-Curie IF grant agreement No. 101023175; and NF-C by a contract of the Centro de Investigación Biomédica en Red en Enfermedades Raras (CIBERER), an initiative of the ISCIII (Spain).
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.