
Abstract
Background: Cortical spreading depolarization (CSD) has been implicated in the pathophysiology of migraine with aura. Patients that suffer from this type of migraine have shown a higher risk of developing an ischaemic stroke.
Case: A 42-year-old female exhibited reoccurring migraine attacks for the first time 1 month before suffering an ischaemic infarction. Imaging studies revealed an occlusion in the right middle cerebral artery. Other possible disorders were excluded. It was possible to register 20 CSDs, of which 12 coincided with high levels of glutamate and lactate/pyruvate ratio. Loss of electrocorticographic activity was observed for 89 hours after the 8th depolarization.
Conclusions: Migraine with aura symptoms may be induced by CSDs triggered by hypoperfusion states. Our case supports the idea of the migraine with aura–stroke continuum.
Keywords
Introduction
Many investigations have suggested a complex bidirectional relationship between migraine with aura (MA) and ischaemic stroke. Migraine has been identified as an independent risk factor for cerebral ischaemic events, but the relationship between MA and ischaemic stroke remains unclear. Many hypothetical mechanisms have been proposed (1,2). It seems that cortical spreading depolarization (CSD), implicated in the pathophysiology of MA (3), could be involved in this relationship.
We present a case of a young female who suffered from MA attacks prior to a right middle cerebral artery (MCA) infarction and displayed clear-cut spreading depolarizations in perifocal electrocorticographic (ECoG) recordings.
Case presentation
A 42-year-old right-handed woman presented with recurrent headaches, which pulsated and were of moderate to severe intensity, 1 month before admission. They occurred mostly unilaterally and occasionally were associated with nausea. These symptoms were sustained for some hours and sometimes improved by taking aspirin. The patient presented a family history of migraine (mother and daughter), though she had never experienced migraine attacks herself. However, she had a past medical history of hypothyroidism. Risk factors included obesity and nicotine abuse. During the 4 days prior to admission, the headaches were preceded by visual disturbances, scotomas and photopsias lasting for several minutes and nausea.
Seven hours before arriving at a local hospital, the patient presented for the first time with a particular visual symptom, which she described as a big flickering red point in the middle of the ocular field that gradually increased in size. This was followed by a severe throbbing headache, nausea and photophobia. All of the previously described symptoms persisted for nearly 6 hours. She sought help in ambulatory care, was diagnosed with MA, prescribed aspirin and sent back home. Approximately 6 hours after the appearance of the visual aural symptoms, she developed confusion and hemiparesis in the left extremities, which was followed by the sudden presence of complex partial seizures and loss of consciousness.
The patient was sedated and transported to the nearest hospital. Four hours after the onset of convulsions a head CT scan was performed, showing early signs of cerebral ischaemia in the right MCA territory without haemorrhage. In anticipation of a malignant course of MCA infarction, the patient was transferred to our hospital. At the time of admission, her National Institute of Health Stroke Scale (NIHSS) was 19 (somnolent, anarthria, left hemiparesis). An angio-CT study was performed revealing an infarct in the right MCA with occlusion in the M1 segment and compression of the right ventricle. MRI studies documented an infarction of >2/3 vascular territory of the MCA. A right-hemispheric hemicraniectomy with duraplasty was performed ∼25 hours after the first onset of symptoms. The patient was monitored multimodally in the intensive care unit. A parenchymal intracranial pressure (ICP) probe, a thermodiffusion cerebral blood flow (CBF) probe, microdialysis catheter and subdural electrode strip were placed in the right frontal lobe, in a tangential relationship to the border of the infarction (Figure 1A and B).
(A) The position of the ECoG electrodes is shown. (B) Native computed tomography control post trepanation, where a right median infarct on the second day of progression and the position of the ECoG electrodes are shown.
In total, 20 CSDs were observed, which consisted of a slow potential change emerging from the channel nearest to the infarct rim. Most of the CSDs (95%) were recorded within the first 3 days of ECoG monitoring and occurred at intervals of approximately 140 minutes. These presented a mean propagation velocity of 2.7 mm/min (0.94–10 mm/min) and peak-to-peak amplitude of 5 mV (1.4–13 mV). Recovery of the electrical activity was prolonged after successive CSDs. Especially after the 8th spreading depolarization; the most lesioned channel remained without restoration of electrocorticographic activity for 89 hours, eventually undergoing a partial recovery (Figure 2A). A group of 12 consecutive depolarizations, observed 20 hours from the beginning of monitoring, coincided with the increased levels of glutamate and lactate/pyruvate ratio (Figure 2B). No relationship was found between CSD and MAP, ICP, CPP or glucose. The patient was monitored for 6 days and was treated according to ICP/CPP and general measures of stroke neurointensive care. No infarct growth was seen in the follow-up MRI after 6 days. No other casual pathology was displayed. All laboratory tests, as well as blood pressure monitoring, electrocardiography, transesophageal echocardiography and Doppler ultrasonography of the cerebrovascular system were normal. Three weeks after the event, the patient was oriented with dysarthria, dysphasia, left hemiparesis and hypoesthesia. Six months after, she showed an extended GCS of 3 (lower severe disability). Interestingly, the patient has not experienced migraine attacks since the resolution of the ischaemic event.
(A) During the most critical period, more cortical spreading depolarizations (CSDs) were recorded. Examples of CSDs are shown on the left side, using a high-pass filtered ECoG > 0.5. In the 2nd CSD, the depression of electrical activity can be seen on channels 4 and 5. In the 8th CSD, the basal electrical activity is already diminished before the CSD appeared. The recovery is partial and slower. In the 14th CSD, the three channels are involved. (B) Time points where CSDs appeared are indicated with “×”. Changes of glutamate concentration and lactate/pyruvate ratio using brain microdialysis during the monitoring time are shown. There was an acute increase in both markers, compatible with acute cell damage. No clear reason was found to explain such a dramatic and transient change.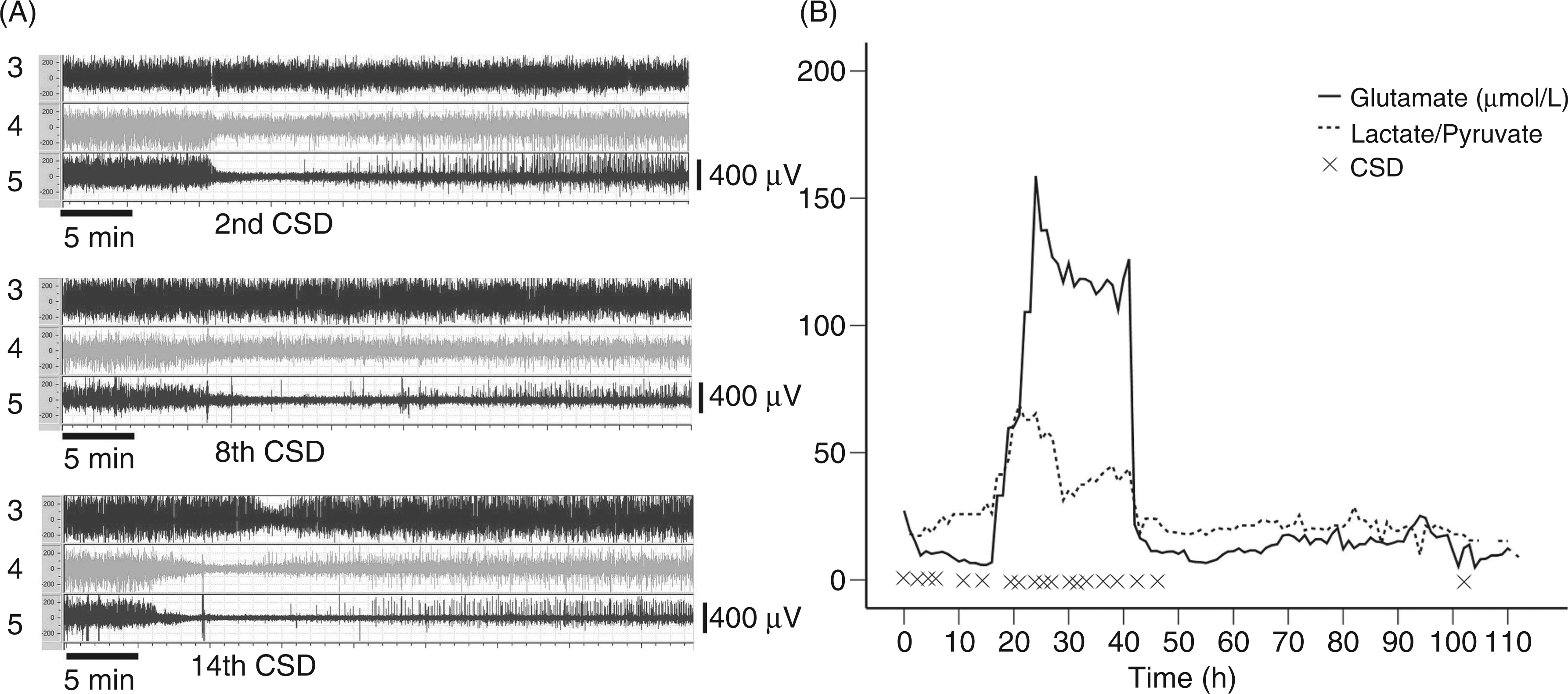
Discussion
The correct categorization of migraine-related stroke syndromes is complex and requires precise details from medical history. Moreover, the delayed admission of patients can hinder the correct diagnosis. Our patient did not fulfil the diagnostic criteria of the International Headache Society (IHS) for migrainous infarction (4). Despite the fact that the cause of cerebrovascular occlusion remains enigmatic and no other pathology could be demonstrated, it is probable that thromboembolism was the origin. Therefore, a diagnosis of cerebral infarction with a different cause, presenting symptoms resembling MA, was established.
This case reinforces the hypothesis that hypoxic-ischaemic episodes can induce MA attacks (5), where the most likely explanation underlying the induction of migraine visual aura is CSD (3). This is supported by different imaging studies, which show that changes resembling CSD are present during aura symptoms. Consequently, such symptoms may develop when the slow depolarization waves expand through the occipital visual lobe (6). CSDs also explain migraine headache, by activating the trigeminovascular reflex through meningeal inflammation (7). In this regard, recent experimental studies in animals have shown that endothelin-1-induced vasospasm and microembolism may be a link between CSDs and MA (8,9). According to this view, migraine and stroke may both be induced by a continuum of vascular complications capable of triggering CSDs, such as hypoperfusion disorders, vessel dysfunction, release of vasoactive substances, platelet hyperactivity and paradoxical embolism. Moreover, endothelial dysfunction may be a predisposition to ischaemic stroke, by means of vascular reactivity impairment, platelet activation or by facilitating the inflammatory process (10).
In this case, due to the clinical presentation of the patient, we speculate that the possible sequences of events were as follows: there was a predisposition to endothelial dysfunction and gradual narrowing of the right MCA territory. Subsequently, a hypoperfusion state developed that triggered spreading depolarizations accompanied by spreading ischaemia, causing a more severe vasoconstriction with a higher decrease of CBF (11). As a consequence, more CSDs were generated, activating the trigeminal meningeal afferents, which triggered the headache episodes (7). The continuing release of vasoconstrictors during this period resulted in vasospasm, oedema and necrosis, which increased the perfusion deficit (11). The hypoxic state was enough for stronger CSDs capable of invading the visual cortex to be induced, giving rise to the visual aura symptoms. Therefore, a continuum of CSDs, MA and hypoperfusion was established. It is probable that the intense reduction of CBF enabled turbulence and the subsequent stagnation of blood flow, severe enough for a thrombosis to develop, occluding the M1-segment in the right MCA, causing the ischaemic stroke. After the ischaemic infarction due to vascular occlusion, the penumbra area continued developing CSDs, which were registered by the ECoG.
The exact chain of events remains enigmatic and there is no way to prove this in retrospect. However, such a scenario was previously described by Olesen et al. (5) in patients with carotid artery stenosis, showing that thromboembolic events can trigger typical MA attacks with or without headache, particularly in migraineurs and borderline ischaemia, which lowers the threshold for developing MA, resulting in a flurry of daily attacks that can last several weeks. They conclude that ischaemia-induced migraine attacks may be more frequent than migraine-induced ischaemic insults. There is also an interesting case reported by Klingebiel et al. (12) showing a patient with high grade internal carotid artery stenosis and scattered focal laminar cortical infarcts, who developed a status aurae migraenalis. This was abolished after thrombendarterectomy of the stenosed internal carotid artery; the repetitive visual spreading scintillation scotoma. They suggest a cycle, where perfusion deficit has induced CSDs that generate cortical lesions by microarterial spasm. In consequence, CSD propagation caused the clinical visual symptoms, which are perpetuated as long as the perfusion deficit exists.
Phenomenologically, the CSDs observed in this patient displayed similar characteristics to those reported in stroke patients. This was true in terms of the absolute number of depolarizations, morphology and spread velocity (13). Finally, an increase in extracellular concentrations of glutamate and lactate/pyruvate ratio was observed, in association with a CSD cluster. Regional metabolic dysfunction has been suggested as a predictor of poor outcome; in particular, increments in lactate/pyruvate ratio are associated with cell hypoxia and imbalance between metabolic rate and oxygen supply. At the same time, glutamate is known to generate and propagate CSDs in animal models and is an indicator of compromised brain metabolism and acute neuronal damage (14).
In conclusion, MA symptoms may be triggered by hypoxic-ischaemic events. Our case supports the idea of the migraine with aura–stroke continuum.
Footnotes
Acknowledgements
We gratefully acknowledge the contributions by the medical and nursing staff of the neurosurgical intensive care units of Heidelberg.
Funding
This work was supported by a grant from the ZNS – Hannelore Kohl Stiftung (2004006).
Conflict of interest
None declared.