
Abstract
Objective
The neuropeptide calcitonin gene-related peptide (CGRP) has now been established as a key player in migraine. However, the mechanisms underlying the reported elevation of CGRP in the serum and cerebrospinal fluid of some migraineurs are not known. A candidate mechanism is cortical spreading depression (CSD), which is associated with migraine with aura and traumatic brain injury. The aim of this study was to investigate whether CGRP gene expression may be induced by experimental CSD in the rat cerebral cortex.
Methods
CSD was induced by topical application of KCl and monitored using electrophysiological methods. Quantitative PCR and ELISA were used to measure CGRP mRNA and peptide levels in discrete ipsilateral and contralateral cortical regions of the rat brain 24 hours following CSD events and compared with sham treatments.
Results
The data show that multiple, but not single, CSD events significantly increase CGRP mRNA levels at 24 hours post-CSD in the ipsilateral rat cerebral cortex. Increased CGRP was observed in the ipsilateral frontal, motor, somatosensory, and visual cortices, but not the cingulate cortex, or contralateral cortices. CSD also induced CGRP peptide expression in the ipsilateral, but not contralateral, cortex.
Conclusions
Repeated CSD provides a mechanism for prolonged elevation of CGRP in the cerebral cortex, which may contribute to migraine and post-traumatic headache.
Introduction
Migraine is a complex, multifactorial neurological disorder that is conservatively estimated to affect ∼12% of Americans (1). The most prominent characteristic of migraine is the disabling headache, which manifests as a throbbing, unilateral pain made worse with routine activity, and coincident with nausea/vomiting and/or photophobia/phonophobia. Approximately one-third of migraineurs experience a premonitory aura, which typically manifests as a disruption in the ipsilateral visual hemifield (2). The pathophysiological substrate of the visual aura is cortical spreading depression (CSD), a transient wave of neuronal and glial depolarization, followed by a sustained depression of electrical activity (3,4). CSD is associated with a massive translocation of ions and release of nitric oxide, arachidonic acid, glutamate, and ATP (5,6). The sudden rise in extracellular K+, arachidonic acid and nitric oxide is the likely trigger for CSD-induced activity in meningeal nociceptors (7) and central trigeminovascular neurons (8). CSD may also lead to migraine by potentiation of an inflammatory response in the dura (9).
In addition to the connection between migraine and CSD, it is well accepted that CSD also occurs following acute brain injuries, such as traumatic brain injury (TBI) and strokes (10–12). Whereas the pattern of brain injury-triggered CSD is heterogeneous and influenced by many factors, a common feature is that there are multiple CSD events, often about every 30 minutes for many hours to days (11). For example, 72% of subarachnoid hemorrhage patients experience clusters of repetitive CSD events (13), and 56% of TBI patients experience repeated spreading depression events (mostly CSD), with a total of 1328 events observed in 58 patients over 67 hours (14). Hence, brain injury in humans can lead to tens to hundreds of CSD events over days. Repeated CSD events have also been observed for hours to days in some, but not all, animal TBI models (12).
It has been shown that multiple CSD events can modulate many genes at early (hours) and late (days to weeks) time points across a variety of gene ontologies (15,16). A potential candidate for regulation by CSD is calcitonin gene-related peptide (CGRP). CGRP is a vasoactive neuropeptide that is widely distributed in the central and peripheral nervous systems. Clinical and preclinical studies have established CGRP as a key player in migraine (17). Intravenous CGRP administration to migraineurs is sufficient to elicit a migraine-like headache (18,19), and CGRP levels have been reported to be elevated in both the serum and CSF of migraineurs (20). Importantly, CGRP receptor antagonists and CGRP-blocking antibodies can ameliorate migraine symptoms (21,22).
In this report, we have asked whether CSD is sufficient to alter CGRP expression. Such a link would fit in the context of interesting, but limited, evidence of CGRP involvement in CSD (17,23). In particular, a calcium-dependent release of CGRP was observed during CSD and inhibition of CGRP receptors reduced the magnitude of CSD in rat neocortical slices (24). Elevated CGRP may modulate neurotransmission and possibly contribute to sensory hypersensitivity (17,25). For this scenario, we reasoned that the elevated synthesis, if it occurred, would likely be maintained for a relatively long time (24 hours). Similar prolonged times were also required for activation of CGRP gene expression in trigeminal ganglia organ cultures (26), and by epigenetic reprogramming of glial cells (27). We therefore proposed that CSD might be a mechanism by which cortical levels of CGRP become elevated for a prolonged period in migraine and TBI patients.
Materials and methods
Animals
Adult male Sprague–Dawley rats (n = 36, 240–440 g; Shanghai SLAC Laboratory Animal Corporation, Ltd.) with food and water ad libitum were used. Experimental procedures were performed in the animal unit of Soochow University. All rats were healthy, with no drug treatments or previous tests carried out prior to experimentation. Animals were housed two per cage in specific pathogen-free conditions with standard bedding material and rat special synthetic feed. Animals were allowed to acclimate to the housing room for 7 days prior to the experiment, then matched by body weight into experimental groups of sham and CSD, which were performed randomly on different days using one rat per day at various times during the day. No animals were excluded from the analysis. During the experiment, rats were given isoflurane anesthesia, which was monitored by absence of whisker movements and lack of reaction to brief tail pinches. After surgeries, animals were given an antibiotic and anti-inflammatory, as described below, and outwardly appeared healthy. Euthanasia was by excess isoflurane exposure. All procedures were approved by the Ethic Review Panel of Soochow University under agreement with XJTLU, and performed in accordance with Chinese national guidelines and in adherence to ARRIVE guidelines.
CSD induction
Animals were anesthetized with isoflurane (5% induction, 2.5–3.5% during surgery, 1.0–1.5% maintenance) in O2: N2O (1:2). A small incision was made and two small burr holes were drilled carefully in the skull right frontoparietal region (1 mm i.d.; Figure 1). The posterior hole (4 mm posterior, 2 mm lateral to bregma) was prepared with extra care to minimize damage to the underlying dura. A silver chloride recording electrode was implanted through the anterior hole (3 mm anterior, 2 mm lateral to bregma) for recording of CSD waves. A reference electrode was placed under the scalp. Both holes were moisturized with artificial cerebrospinal fluid (ACSF; 125 mM NaCl, 2.5 mM KCl, 1.18 mM MgCl2, 1.26 mM CaCl2, pH 7.3). Rectal temperature was maintained at 37℃. Upon completion of surgery, rats were maintained under anesthesia for at least 1 hour to allow for stabilization and tissue recovery.
Cortical spreading depression (CSD) induction and propagation in the rat cortex. (A) CSD was induced with topical application of 1 µl 3 M KCl (or artificial cerebrospinal fluid for sham) onto the dura via a posterior burr hole. An anterior hole was used for CSD recording. A total of 36 rats were used. Of these, 26 rats were used for multiple CSD experiments including 14 for CSD induction and 12 for sham. To minimize the animal use, three of 14 rats in the CSD group and three of 12 in the sham group were also used for measuring calcitonin gene-related peptide (CGRP) levels in addition to CGRP mRNA. In the single CSD group, 10 rats were used with five for CSD and sham, respectively. (B) A representative trace showing CSD propagation and magnitude (indicated as area under the curve, AUC, grey lines).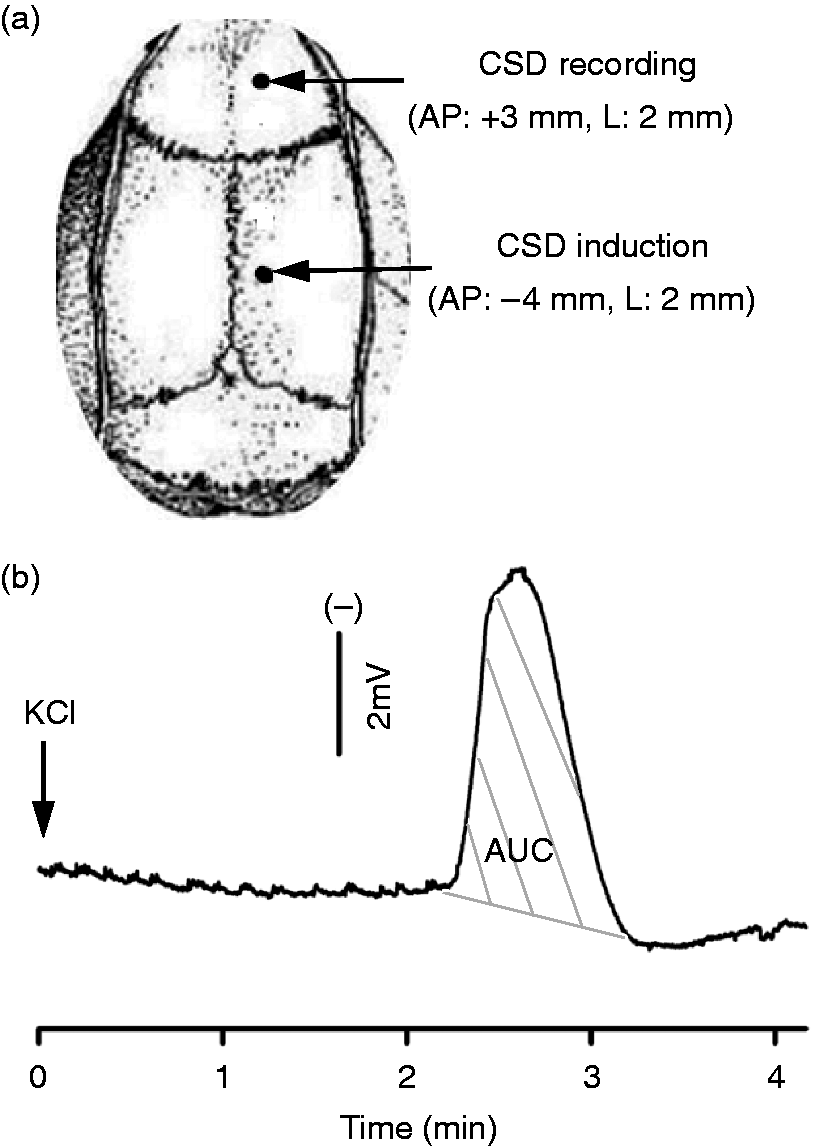
For the multiple CSD group (n = 14), 1 µl of 3 M KCl was carefully dropped into the posterior hole for single CSD wave elicitation. As soon as the first CSD wave was detected at the recording site, KCl was removed using a tissue, followed by washout with ACSF and placement of ACSF-moistened cotton over the hole. For the sham group (n = 12), 1 µl ACSF was used. In both groups, five repeated CSD or sham episodes were elicited at 40-minute intervals. Both electroencephalogram (to monitor depth of anesthesia) and direct current potentials (to monitor CSD events) were recorded and analyzed using LabVIEW. The electrode was removed and wound sutured after the fifth CSD episode. Ibuprofen (5–10 mg) and mupirocin ointment (0.4–0.8 mg) were applied. At 24 hours after the fifth CSD episode, the rat was re-anesthetized for euthanasia and tissue removal.
For the single CSD group (n = 5), CSD induction, CSD recording, and post-surgery care were the same as that for the repeated CSD, except that only one CSD episode was elicited by 1 µl of 3 M KCl. For the sham group (n = 5), 1 µl ACSF was used. At 24 hours, the rat was re-anesthetized for euthanasia and tissue removal.
qPCR
All quantitative polymerase chain reaction (qPCR) assays on multiple CSD tissue samples were performed blinded at the University of Iowa using coded samples shipped from XJTLU. Some of those samples were also tested at XJTLU as an internal control for measurements on the single CSD samples, which were done at XJTLU. In both locations, the same protocols and reagents were used. Whole cortex or cortical regions (50–100 mg) were homogenized in 1 ml TRIzol (Sigma-Aldrich) and RNA concentration and purity measured by Nano-Drop (Thermo Scientific); 1 µg total RNA was reverse transcribed into cDNA using the GoScript™ RT system (Promega). qPCR was performed in duplicate using 1/20th of the cDNA reaction on a Bio-Rad CFX Connect using SYBR-Green Master Mix (Takara Clontech), except for 18S rRNA, which used 1/20,000th of the cDNA. Primers (forward, reverse) were: CGRP (NM_001033953.2) 5′AACCTTGGAAAGCAGCCCAGGCATG3′, 5′GTGGGCACAAAGTTGTCCTTCACCA3′; and three reference genes: peptidylprolyl isomerase A (PPIA) (NM_017101.1) 5′TTGCTGCAGACATGGTCAAC3′, 5′TGTCTGCAAACAGCTCGAAG3′; β-actin (ACTB) (NM_001101.3) 5′ACGGTCAGGTCATCACTATGG3′, 5′AGCCACCAATCCACACAG3′; and 18S rRNA (NR_046237.1) 5′ATGGCCGTTCTTAGTTGGTG3′, 5′AACGCCACTTGTCCCTCTAA3′. All qPCR data from multiple CSD samples were analyzed using both absolute quantification of CGRP mRNA (with standard curves) and relative fold change (2−ΔΔCq method) normalized to each contralateral hemisphere. The single CSD samples were analyzed only by the relative fold change (2−ΔΔCq method) normalized to the sham tissue. Standard curves were generated using plasmids containing PCR products in pCR2.1 (Invitrogen) (confirmed by sequence). CGRP mRNA levels were normalized to the product-based geometric mean of the three reference genes (28), calculated as the cube-root of the product of the reference genes (PPIA × β-actin × 18S)1/3 divided by 10,000. Similar results were observed when CGRP levels were normalized to each individual reference gene. CGRP levels are mean ± SEM.
ELISA
Protein extraction and detection from cortex homogenates followed the manufacturer’s instructions using the rat CGRP enzyme-linked immunosorbent assay (ELISA) kit (Bertin Pharma). To minimize the number of animals, cortical regions were snap frozen, pulverized, and split to allow both protein and RNA extractions. Due to the small amounts of tissue, it was necessary to combine motor, somatosensory, and visual cortices for protein, and set aside cingulate and frontal cortices for only RNA. All protein samples were rapidly homogenized within 15 seconds in 2 N acetic acid at 2 ml/100 mg tissue, then heated at 90℃ for 10 minutes, centrifuged at 10,000 g for 30 minutes, dried for 1 hour, and stored at −80℃. Immediately before assay, samples were reconstituted with enzyme immunoassay (EIA) buffer, and analyzed using a microplate reader. CGRP levels are mean ± SEM.
Data analysis
For qPCR data, statistical analyses were performed using GraphPad Prism software as follows: comparisons across groups were done using an ordinary one-way analysis of variance (ANOVA) Kruskal–Wallis test; comparisons between ipsilateral versus contralateral hemispheres within the group were done using one-tailed Wilcoxon –t-test; comparisons between CSD and sham groups were done using one-tailed Mann–Whitney t-test. For ELISA data, normal distribution was confirmed by SPSS 16.0 software, and statistical analyses were performed using GraphPad Prism software with comparisons across groups using one-way ANOVA; comparisons between ipsilateral versus contralateral hemisphere within the group using one-tailed paired t-test; comparisons between CSD and sham groups using one-tailed unpaired t-test.
Results
Detection of experimentally induced CSD in rats
In the sham group, ACSF administration was insufficient to elicit CSD. In the CSD group, 1 µl of topical 3 M KCl onto the dura resulted in a CSD wave that began ∼2–3 minutes after administration (Figures 1A and 1B). CSD propagation was identified by a transient, negative shift of the direct current–potential, which was observed in all CSD rats monitored by this means. For the rats that underwent multiple CSD events, the magnitude of each event was approximately 9 mV × minutes and there was no significant difference in the number or magnitude of CSD episodes over the 5 KCl applications (p = 0.89) (not shown). When summed up, the mean accumulative magnitude of CSD for each rat was 53.0 ± 10.2 mV × minutes in the multiple CSD group (n = 14) and 5.9 ± 1.8 mV × minutes (n = 5) in the single CSD group. Generally, only a single CSD wave was observed after each of the applications; although in four rats a second wave was observed after one of the applications in the multiple CSD group.
Increased cortical CGRP gene expression at 24 hours post-multiple CSD
In the sham group, the copy number of contralateral and ipsilateral CGRP mRNA was 13.5 ± 2.9 and 14.1 ± 2.2, respectively (p = 0.69), indicating that the surgical procedure did not significantly change CGRP gene expression (Figure 2A).
Ipsilateral calcitonin gene-related peptide (CGRP) mRNA is upregulated 24 hours post-multiple cortical spreading depression (CSD). (a) Absolute levels of CGRP mRNA were significantly elevated in the ipsilateral (ipsi), but not contralateral (contra), cortex post-CSD. There was no significant increase in the sham-treated rats. Data for individual rats are shown with lines connecting the paired cortices. (b) Comparison of CGRP mRNA levels between contralateral and ipsilateral cortices shown for individual rats (left panel) and as the fold change (right panel). Increased relative expression of CGRP mRNA in the ipsilateral normalized to contralateral cortex. In both (a) and (b), sham (n = 6), CSD (n = 8), **p < 0.01; ***p < 0.001.
In the CSD group, the copy number of CGRP mRNA in the contralateral hemisphere was 18.6 ± 2.2, which is not significantly different from that of contralateral hemisphere in sham rats (p = 0.14; Figure 2A), demonstrating that repeated unilateral CSD does not affect CGRP gene expression in the contralateral hemisphere. In contrast, repeated CSD events in the ipsilateral hemisphere significantly increased the CGRP mRNA copy number to 79.1 ± 22.7 at 24 hours post-CSD (p < 0.001; Figure 2A). Comparison of relative ipsilateral CGRP levels normalized to the contralateral hemisphere of each rat agrees with the measured absolute levels. The sham group showed a 1.3 ± 0.4-fold increase between hemispheres, whereas the CSD group had a significantly greater 3.8 ± 0.8-fold increase (p = 0.004; Figure 2B).
No increased CGRP gene expression at 24 hours post-single CSD
In the single CSD group, CSD ipsilateral cortex did not show a significant change in CGRP mRNA levels. There was a 1.28 ± 0.06-fold change compared with that in sham ipsilateral group (p = 0.11; Figure 3). Contralateral samples were not included in this test, as it was shown that surgery did not significantly alter CGRP gene expression during the multiple CSD tests (Figure 2). Thus, it is very unlikely an elevation of CGRP gene expression in the contralateral cortex would be observed with single CSD.
Ipsilateral calcitonin gene-related peptide (CGRP) mRNA is not altered 24 hours post-single cortical spreading depression (CSD). Comparison of CGRP levels between ipsilateral (ipsi) cortices of CSD rats (n = 5) normalized to sham rats (n = 5). There was no significant difference (p = 0.11).
Regional induction of CGRP in the cortex
We then examined CGRP mRNA levels in discrete regions of the cortex. In the sham group, CGRP mRNA levels in the contralateral and ipsilateral frontal, motor, somatosensory, and visual cortices were similar (Figure 4A). The cingulate had slightly higher CGRP mRNA levels in the sham contralateral (16.7 ± 4.0) and ipsilateral (32.9 ± 12.0) cortices (Figure 4A), which is consistent with a previous report that the rat cingulate has elevated CGRP relative to other cortical regions (29). There was no significant change in CGRP mRNA content between contralateral and ipsilateral hemispheres of sham frontal, cingulate, or somatosensory cortices (p = 0.16, 0.11, and 0.22, in respective order), indicating that the surgery does not alter CGRP mRNA expression in these regions. However, a slight, but significant, increase in CGRP mRNA was observed in the motor and visual cortices of sham rats (p = 0.016 and 0.031, respectively; Figure 4A).
Multiple cortical spreading depression (CSD)-induced calcitonin gene-related peptide (CGRP) mRNA expression shows regional specificity. (a) Top view of rat brain cortical regions used for dissections. Cortical regions are designated as 1 = frontal, 2 = cingulate, 3 = motor, 4 = somatosensory, and 5 = visual. The CSD recording and induction sites are also indicated. (b) Absolute levels of CGRP mRNA were significantly elevated in ipsilateral (ipsi) frontal, motor, somatosensory (SS), and visual cortices, but not in the cingulate (Cing) cortex, or the contralateral (contra) hemispheres, 24 hours post-multiple CSD. (c) Significant increases in relative ipsilateral CGRP mRNA were observed in the frontal, motor, somatosensory, and visual cortices, but not in the cingulate cortex, at 24 hours post-multiple CSD. In both panels, sham (n = 6), CSD (n = 6), *p < 0.05; **p < 0.01; ***p < 0.001.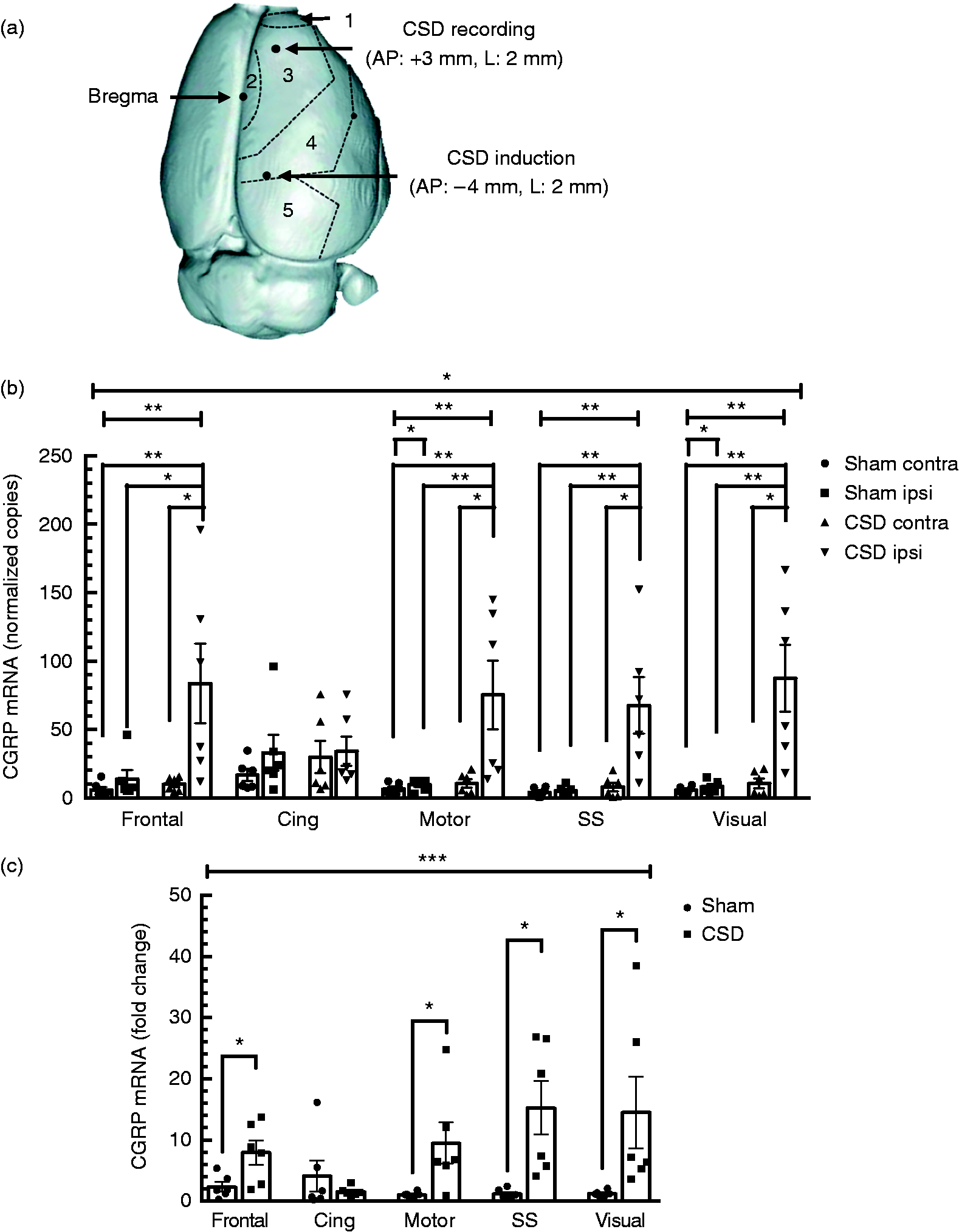
Following CSD, CGRP mRNA levels were significantly increased in the ipsilateral frontal, motor, somatosensory, and visual cortices when compared with sham (p = 0.013, 0.001, 0.001, and 0.001, respectively; Figure 4A). In contrast, CSD did not alter CGRP mRNA levels in the ipsilateral cingulate cortex compared with sham (p = 0.45; Figure 4A). As with the whole cortex, comparison between CSD and sham cohorts of the relative CGRP levels normalized to the contralateral hemisphere of each rat agreed with the absolute levels. In the CSD rats, there were significant increases in the ipsilateral compared with contralateral hemispheres of the frontal cortex (8.0 ± 1.8, p = 0.013), motor cortex (9.5 ± 3.1, p = 0.013), somatosensory cortex (15.3 ± 4.0, p = 0.001), and visual (14.5 ± 5.4, p = 0.001) compared with sham rats (Figure 4B). Also, as seen with absolute expression data, the normalized cingulate cortex did not exhibit a CSD-induced increase over sham (1.6 ± 0.3, p = 0.45; Figure 4B).
Increased CGRP peptide expression at 24 hours post-multiple CSD
Aliquots of somatosensory, motor, and visual cortices used for RNA analyses were combined and used for parallel peptide measurements (see Methods). In the sham group, CGRP peptide levels were 1.4 ± 0.5 ng/g tissue in the ipsilateral hemisphere and 1.8 ± 0.1 ng/g tissue in contralateral (p = 0.27), indicating that surgical procedures do not alter overall CGRP expression under these conditions (Figure 4). In the CSD ipsilateral cortex, the CGRP level was 4.0 ± 0.6 ng/g tissue, which was about 2.9-fold higher than sham ipsilateral cortex (p = 0.014; Figure 5).
Ipsilateral calcitonin gene-related peptide (CGRP) peptide levels are upregulated post-multiple cortical spreading depression (CSD). CGRP peptide levels are significantly elevated in ipsilateral (ipsi) cortex 24 hours post-CSD (n = 3), but not the contralateral (contra) hemispheres, nor in sham rats (n = 3), *p < 0.05.
Discussion
In this study, we have shown that unilateral multiple CSD is sufficient to upregulate CGRP mRNA and peptide levels in the ipsilateral cerebral cortex. The RNA induction was widespread across much of the cerebral cortex: in the frontal, motor, somatosensory, and visual cortices, but not in the cingulate cortex. The lack of induction in the cingulate is consistent with reports that CSD propagation is less efficient in this region (30). Importantly, the fact that CGRP mRNA levels were similarly increased in multiple cortical regions indicates that the elevated expression is attributed to CSD, rather than depolarization in the immediate area of KCl application. These data extend an earlier in vitro study showing that CSD induced CGRP release in rat neocortical slices (24). Thus, CSD might contribute to the elevated CGRP in the CSF of some migraine patients. However, it is very unlikely that cortical CGRP contributes to the elevation of CGRP in the external jugular vein during migraine. Indeed, Piper et al. (31) clearly documented that CGRP release into the external jugular vein was not increased by experimental CSD in the cat. Whether CSD increases synthesis in the trigeminal ganglia remains to be determined. A recent study reported that CSD increases the number of CGRP-positive cells in rat trigeminal ganglia (32), which may point to increased synthesis. Overall, these data provide evidence linking CSD and CGRP expression that may contribute to migraine pathogenesis.
A key finding of our study was that multiple CSD events were required for robust induction of the CGRP gene. The experimental protocol we used was designed to elicit multiple CSD events similar to that used in other laboratories (9,23,33–35), but which also likely differs from human migraine. Indeed migraine aura is believed to involve only a single CSD event. On the other hand, TBI is commonly associated with up to hundreds of CSD waves that can occur over days following the injury (10–12). To our knowledge, the possibility that individuals with a history of head trauma and spreading depression exhibit elevated CGRP levels has not been investigated. However, in a recent study, Elliott and colleagues observed a sustained elevation of CGRP in the brainstem (most likely from the trigeminal nerve) for at least 4 weeks in rodents exposed to controlled cortical impact injury (36). This injury can cause one to four CSD events over several hours (37). More limited studies have also reported elevated CGRP in other rodent TBI models (38,39). Whether from TBI or migraine, there is increasing evidence that CSD affects behavior and likely potentiates nociception that may in part involve CGRP. CSD activates meningeal nociceptors and central trigeminovascular neurons (7,8,40), and an immediate effect of CSD is reduced movement and freezing responses (41,42). Nonetheless, a limitation of animal CSD studies remains extrapolation to humans, especially as CSD propagation is limited by prominent sulci in the human brain that are absent in the lissencephalic rodent brain. Within this limitation, we predict that TBI may increase CGRP levels to alter brain plasticity and predispose patients to migraine-like post-traumatic headaches.
How might CSD increase cortical CGRP gene expression? While speculative, one mechanism may be generation of reactive oxygen species (ROS). Migraineurs have elevated plasma levels of a ROS-induced lipid peroxidation products (43). In rats, CSD produces ROS in the cortex and trigeminal nerve (44,45). Likewise, the ROS-responsive COX2 gene has been reported to be upregulated by CSD (16). Moreover, ROS-induced CGRP gene expression can be inhibited with antioxidant treatment in rat trigeminal ganglia (26). In light of these observations, we speculate that CSD-induced ROS production can lead to pro-inflammatory cascades that upregulate the CGRP gene. Interestingly, a recent analysis of the literature concluded that oxidative stress is a shared feature of most migraine triggers (43).
We have provided evidence showing that CSD upregulates CGRP gene expression in the cortex. Given the promising potential of CGRP-based therapeutics for treating and preventing migraine, a link between CSD and CGRP further emphasizes the importance of CSD as a target for migraine drug development (46,47). The significance of our finding is that CSD may be a mechanism by which CGRP levels become elevated for a prolonged period in some migraine patients. While there are multiple mechanisms that can potentially increase CGRP expression, including nitric oxide (48) and cytokines (33,48,49), this is the first report of a physiological in vivo mechanism that may elevate cortical CGRP gene expression in migraine and post-traumatic headache. Given CGRP’s role as a neuromodulator (17), this elevation may potentially contribute to cortical hyperexcitability and sensory abnormalities in migraine.
Article highlights
Multiple CSD events can trigger CGRP gene expression in discrete regions of the rat cerebral cortex. This is the first in vivo evidence for a mechanism to initiate and maintain elevated CGRP levels in migraine and post-traumatic headache.
Footnotes
Acknowledgements
We are grateful for advice and training from Dan Kaufmann and KC Brennan (University of Utah) and advice from Sajedeh Eftekhari (UCLA) and Karin Warfvinge and Lars Edvinsson (Lund University). We also thank Liwen Jiang (XJTLU) for technical support on single CSD experiments.
Declaration of conflicting interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: Wangwenli Charitable Foundation (RD0006), NIH (NS075599), Veterans Affairs Medical Center (1IO1RX002101), and Department of Defense USAMRAA (W81XWH-16-1-0071).