
Abstract
Background
Acute and persistent post-traumatic headache are often debilitating consequences of traumatic brain injury. Underlying physiological mechanisms of post-traumatic headache and its persistence remain unknown, and there are currently no approved therapies for these conditions. Post-traumatic headache often presents with a migraine-like phenotype. As calcitonin-gene related peptide promotes migraine headache, we explored the efficacy and timing of intervention with an anti- calcitonin-gene related peptide monoclonal antibody in novel preclinical models of acute post-traumatic headache and persistent post-traumatic headache following a mild traumatic brain injury event in mice.
Methods
Male, C57Bl/6 J mice received a sham procedure or mild traumatic brain injury resulting from a weight drop that allowed free head rotation while under minimal anesthesia. Periorbital and hindpaw tactile stimulation were used to assess mild traumatic brain injury-induced cutaneous allodynia. Two weeks after the injury, mice were challenged with stress, a common aggravator of migraine and post-traumatic headache, by exposure to bright lights (i.e. bright light stress) and cutaneous allodynia was measured hourly for 5 hours. A murine anti- calcitonin-gene related peptide monoclonal antibody was administered after mild traumatic brain injury at different time points to allow evaluation of the consequences of either early and sustained calcitonin-gene related peptide sequestration or late administration only prior to bright light stress.
Results
Mice with mild traumatic brain injury, but not a sham procedure, exhibited both periorbital and hindpaw cutaneous allodynia that resolved by post-injury day 13. Following resolution of injury-induced cutaneous allodynia, exposure to bright light stress re-instated periorbital and hindpaw cutaneous allodynia in injured, but not sham mice. Repeated administration of anti-calcitonin-gene related peptide monoclonal antibody at 2 hours, 7 and 14 days post mild traumatic brain injury significantly attenuated the expression of cutaneous allodynia when evaluated over the 14-day post injury time course and also prevented bright light stress-induced cutaneous allodynia in injured mice. Administration of anti-calcitonin-gene related peptide monoclonal antibody only at 2 hours and 7 days after mild traumatic brain injury blocked injury-induced cutaneous allodynia and partially prevented bright light stress-induced cutaneous allodynia. A single administration of anti-calcitonin-gene related peptide monoclonal antibody after the resolution of the peak injury-induced cutaneous allodynia, but prior to bright light stress challenge, did not prevent bright light stress-induced cutaneous allodynia.
Conclusions
We used a clinically relevant mild traumatic brain injury event in mice along with a provocative stimulus as novel models of acute post-traumatic headache and persistent post-traumatic headache. Following mild traumatic brain injury, mice demonstrated transient periorbital and hindpaw cutaneous allodynia suggestive of post-traumatic headache-related pain and establishment of central sensitization. Following resolution of injury-induced cutaneous allodynia, exposure to bright light stress re-established cutaneous allodynia, suggestive of persistent post-traumatic headache-related pain. Continuous early sequestration of calcitonin-gene related peptide prevented both acute post-traumatic headache and persistent post-traumatic headache. In contrast, delayed anti-calcitonin-gene related peptide monoclonal antibody treatment following establishment of central sensitization was ineffective in preventing persistent post-traumatic headache. These observations suggest that mechanisms involving calcitonin-gene related peptide underlie the expression of acute post-traumatic headache, and drive the development of central sensitization, increasing vulnerability to headache triggers and promoting persistent post-traumatic headache. Early and continuous calcitonin-gene related peptide blockade following mild traumatic brain injury may represent a viable treatment option for post-traumatic headache and for the prevention of post-traumatic headache persistence.
Abbreviations
Cutaneous allodynia Calcitonin gene-related peptide Mild traumatic brain injury Post-traumatic headache Acute post-traumatic headache Persistent post-traumatic headache
Keywords
Introduction
Post-traumatic headache (PTH) is one of the hallmark symptoms following mild traumatic brain injury (mTBI), often presenting with a migraine-like phenotype (1). There are over 2.5 million TBI-related visits yearly to hospital Emergency Departments (2), and an overabundance of unreported head injuries in the United States annually (3,4). It is estimated that 70–90% of these injuries are considered “mild” (5). The most common causes of TBI include falls, motor vehicle collisions, being struck, and sports (6–8). In addition, it is estimated that upwards of 20% of US military service members serving in Iraq and Afghanistan suffer from head-related injuries (9), of which 80% can be classified as mTBI (10). Counter-intuitively, greater prevalence and duration of PTH is more commonly associated with mTBI as opposed to more severe TBI (1,11–13). Furthermore, patients who report headache amongst their initial symptoms of mTBI have a higher likelihood of having persistent symptoms than individuals who do not (14,15). While PTH typically resolves over the first weeks to months following mTBI, 15–53% of individuals continue to experience persistent PTH (PPTH) a year after mTBI (16,17).
The underlying pathophysiology driving PPTH remains unknown. PTH may be continuous and unrelenting, or may consist of pain episodes that are associated with triggering events including those commonly reported with migraine such as stress, exercise, sleep disruption and others (18). Pre-existing headache/migraine may be a factor in increased risk of development of PPTH following mTBI (13,19,20). It is not known if the frequency of PTH attacks represents an additional risk promoting the development of PPTH. Extensive evidence from clinical and preclinical studies has demonstrated a pivotal role of CGRP in the pathophysiology of migraine (See reviews by Tepper (21) and Edvinsson (22)), and CGRP has also been implicated in PTH (23,24). Preclinical rodent models of migraine and medication overuse headache have demonstrated increased CGRP in the jugular blood (25,26), a factor that appears causal to pain behaviors as administration of an anti-CGRP monoclonal antibody (mAb) prevents cephalic and extracephalic cutaneous allodynia (CA) (27).
The aim of this study was to investigate the role of CGRP in the development of acute PTH (APTH) and in promoting PPTH following mTBI. As PTH is a secondary headache, it may be effectively modeled in preclinical settings. We adapted a clinically relevant mTBI model via a weight drop in healthy mice that effectively reproduces many of the biomechanics associated with the injury, including unrestrained head impact with linear and rotational acceleration (28–31). This method has been previously demonstrated in rodents (both mice and rats) to produce no observable radiologic, gross or histological brain damage, no skull fractures, minimal loss of consciousness, and no neurological deficits (28,29), similar to what is observed in humans following mTBI. As stress and light are common migraine triggers, mice were exposed to a period of bright light stress (BLS) following resolution of initial mTBI-induced cutaneous allodynia. In order to provide continuous sequestration of CGRP after mTBI, animals received either early and repeated administration of an anti-CGRP mAb, consistent with its calculated half-life in mice, or a control isotype monoclonal antibody. In a separate experiment, anti-CGRP mAb was administered after resolution of mTBI-induced CA but before BLS. We hypothesized that mTBI-induced CGRP promotes acute pain relevant to APTH as well as central sensitization characterized by sustained vulnerability to triggering events, including stress, resulting in pain behaviors consistent with a persistent state of PPTH. Blockade of CGRP following mTBI may therefore prevent the establishment of a sensitized state, inhibiting both the acute phase of PTH and PTH persistence.
Materials and methods
Animals
Male, C57Bl/6 J adult mice weighing 17–22 grams were housed five to a cage on a 14/10-hour light/dark cycle (5 am to 7 pm lights on) with food and water ad libitum. Experiments were conducted during the light cycle phase. All experiments were performed in accordance with the ARRIVE reporting guidelines and with the approval of the Mayo Clinic Institutional Animal Care and Use Committee and Teva Pharmaceuticals. A total of 113 mice were used in this study. Group size requirements to obtain significance at the α = 0.05 and statistical power 0.9 were determined from previous experiments using power analysis. Blinding was not possible in these experiments because of visible behavioral impact of mTBI in some mice.
Induction of mild traumatic brain injury
The mouse model of experimental mTBI was adapted from Kane et al. (28). Briefly, mice were lightly anaesthetized with 3% isofluorane and then laid with their ventral surface on an elevated tissue paper stage capable of supporting body weight with the head unrestrained. The paper stage was situated over a plexiglass apparatus with a soft sponge at the bottom. A metal guide tube was directed to the top of the mouse skull between the ears to ensure standardized placement of the weighted drop. The weight (100 g), released from a height of 94 cm, results in a concussive impact to the head, pushing the mouse through the tissue paper and flipping it down to land on the soft sponge. All mTBI mice in this study experienced both rotational and linear head forces. After impact, the weight falls through the apparatus thereby avoiding a second impact with the animal. Following the procedure, the righting reflex was recorded, and mice were returned to their home cages and allowed to recover. Sham animals were anaesthetized and placed on the tissue paper stage but did not undergo the weighted drop or rotational flip. All mice awoke within 5 minutes of the procedure and were observed to confirm that no visual signs of neurological complications arose. Animals remained grouped in their same cohorts following the procedure.
Bright light stress (BLS) challenge
Unrestrained mice were exposed to BLS induced by LED strips (1000 lux output) that were placed on both sides of their home Plexiglass cages for 15 minutes. The parameters of the BLS protocol were modified from our previous studies in Sprague Dawley rats (32) to produce mild stress that arises from endogenous mechanisms and does not elicit significant CA in naïve or sham mice.
Drug administration
Mouse anti-CGRP mAb was provided by Teva Biologics (Redwood City, CA, USA). Based on our previous studies in a rat model of medication overuse headache (27) showing efficacy with an intraperitoneal dose of 30 mg/kg of fremanezumab, a human anti-CGRP antibody, we used the same route and dose of murine anti-CGRP mAb (diluted in 0.9% saline) in the present studies (27). Control animals received intraperitoneal administration of corresponding isotype control antibody, with specific binding to keyhole limpet hemocyanin (KLH) provided by Teva Biologics, or 0.9% saline only.
Behavioral assessment of cutaneous allodynia
Prior to baseline behavioral assessment, mice were placed individually in elevated Plexiglass chambers with mesh flooring, located in a quiet, non-trafficked area and allowed to acclimate for 3 days for 2 hours each day. Starting on day 0 (pre-mTBI baseline) and periodically thereafter, cephalic (periorbital) and extracephalic (hindpaw) allodynia was measured in the same mice following a 2-hour acclimation period. For assessment of periorbital allodynia, a 0.4 g (3.61) von Frey filament was applied 10 times, with just enough pressure to cause the filament to display a slight arch to the periorbital region and a space of 20–30 sec between each application. A positive response was considered to be swiping of the face, shaking of the head, and/or turning away from the stimuli. Running away or rearing up were not considered as positive responses. For assessment of hindpaw allodynia, a 0.6 g (3.84) von Frey filament was applied to the left hindpaw 10 times with just enough pressure to cause the filament to display a slight arch, and a space of 20–30 sec between each application. Sharp withdrawal of the paw, shaking and/or licking the paw were considered a positive response, while lifting of the paw with the filament or running away were not. Frequency response was calculated as ([number of positive responses/10] × 100%). Following mTBI induction, CA was measured for a time course of up to 14 days and in response to anti-CGRP mAb and control antibody/vehicle administration. All behavioral data were collected by the same person (JO).
Assessment of early, repeated anti-CGRP mAb post mTBI on the development of APTH and PPTH
Mice were randomly separated into five groups: mTBI/saline; mTBI/mAb (group A); mTBI/mAb (group B); sham/saline; sham/mAb (see figures for experimental design). After baseline periorbital and hindpaw tactile measurements, mice were lightly anaesthetized with 3% isoflurane and underwent either the sham or mTBI protocol. Two hours post procedure, mice received either anti-CGRP mAb or saline. Assessment of periorbital and hindpaw CA was measured periodically over a time course of 14 days. On days 7 and 14, mice received additional doses of their respective drug treatments, with a noted exception for mTBI/mAb group A, which received anti-CGRP mAb on days 0 and 7 but saline on day 14 while mTBI/mAb group B received anti-CGRP mAb on all three days, 0, 7 and 14. On day 15 or 16 (groups counterbalanced), all mice were exposed to bright light stress for 15 min, after which CA was measured every hour for 5 hours. To emphasize the correlation between acute CA following mTBI (reflecting an acute pain phase) and stress-induced re-instatement of CA (indicative of a persistent phase) in the same group, acute and BLS-provoked data are plotted in one graph.
mTBI produces immediate facial and hindpaw allodynia and delayed, stress-induced allodynia in mice. Tactile response data from all mTBI and sham animals from four separate experiments treated with either saline or control isotype antibody were combined and plotted to determine the average effects. Mechanical periorbital (a) and hindpaw (b) allodynia were measured at baseline (BL) and on indicated days post injury over a time course of 14 days. On day 14, 15 or 16 (depending on the experimental group), when tactile responses were at baseline levels, mice were subjected to 15 minutes of bright light as a stress stimulus (BLS) (yellow line). Periorbital and hindpaw allodynia were measured over a 5-hour time course following BLS. mTBI mice, but not shams, developed acute allodynic responses following mTBI that were re-established after BLS. A two-way ANOVA with Sidak’s multiple comparison test shows significant difference between mTBI and sham mice at times indicated by red asterisk (*). Data are plotted as means and SEM; sham: n = 19 mice; mTBI: n = 39 mice.
Assessment of delayed anti-CGRP mAb administration on the development of PPTH
After pre-procedure CA baselines were measured, mice were lightly anaesthetized and underwent either the sham or mTBI protocol. Assessment of periorbital and hindpaw CA was measured periodically over a time course of 13 days to determine when sensory thresholds returned to pre-mTBI baselines. On day 10, sham and mTBI mice were randomized and received one dose of either anti-CGRP mAb or control antibody. On day 14, all mice were exposed to BLS for 15 minutes, after which CA was measured every hour for 5 hours. The experimental design resulted in a total of four groups: mTBI/control antibody; mTBI/anti-CGRP mAb; sham/control antibody and sham/anti-CGRP mAb. Another cohort of mice was tested using the same mTBI/sham protocol with delayed mAb administration, except that anti-CGRP mAb or control antibody was administered on day 13, 12 hours prior to BLS and allodynia assessment.
Comparison of saline versus control antibody on CA post mTBI and subsequent stress-induced CA
In order to determine that saline and control antibody were equitable control substances, they were directly compared in a separate cohort of mTBI mice. Periorbital and hindpaw CA were measured at baseline and intermittently over a time course of 14 days after mTBI or sham. Two hours, and 7 and 14 days after mTBI, half of the mTBI mice were given intraperitoneal injection of control antibody (30 mg/kg) and the other half were injected with the same volume of saline. At Day 14, all mice were exposed to BLS and tactile sensitivity in the face and hindpaw were recorded hourly for 5 hours.
Anti-CGRP mAb half-life determination
Mouse anti-CGRP mAb was diluted to 5.4 mg/mL in Dulbecco’s phosphate buffered saline (17-512Q, Lonza, Morristown, NJ, USA) and administered (30 mg/kg, i.p.) to male CD-1 mice (Charles River, Wilmington, MA, USA) weighing between 30–50 g. Animals were lightly anaesthetized, and serial 50 µL blood samples were collected at multiple time points by tail snip over a time course of 100 hours. Serum samples were analyzed for CGRP concentrations via a custom ELISA. Briefly, samples were incubated at room temperature for 30 minutes using a biotinylated mouse α-CGRP peptide (H2265, Bachem, Torrance, CA, USA) and immobilized on streptavidin-coated high capacity 96-well plates (15500, ThermoFisher Scientific, Waltham, MA, USA). Serum samples were washed off with an aqueous buffer solution (50 mM Tris pH 7.4, 150 mM NaCl, 0.05% Tween-20, 0.1% BSA). Captured CGRP analytes were complexed to a goat anti-mouse kappa-HRP conjugated secondary antibody (1050-05, Southern Biotech, Birmingham, AL, USA) at room temperature for 30 minutes. The concentration of analyte was quantified via the oxidation of 3,3′,5,5′-Tetramethylbenzidine (TMB) liquid substrate (T0440, Sigma-Aldrich, St. Louis, MO, USA). The oxidation reaction was allowed to proceed for 2 minutes before being quenched by the 1:2 addition of 2 N H2SO4. Absorbance was measured at 450 nm using a FlexStation 3 (Molecular Devices, Sunnyvale, CA, USA).
Data analysis
Data are presented as the mean and standard error of the mean (SEM). All statistical analyses were conducted in Graphpad Prism 7 (GraphPad Software, La Jolla; CA). Two-way repeated measures (RM) analysis of variance (ANOVA) was performed with “time” as the within-subject factor to assess the main effects of time, treatment group, and the interaction between time and treatment on CA. Sidak’s post hoc test for multiple comparisons was used to assess differences between the groups within each time point. Data in Figure 1 comparing two groups (sham and mTBI) are combined from four independent experiments. As the time points used for assessment differed in multiple experiments, data were analyzed using two-way ANOVA with Sidak’s multiple comparisons. Statistical significance was established a priori at 95% (p < 0.05).
The effects of anti-CGRP mAb treatment 2 hours post mTBI on cutaneous allodynia. (a) Injury and treatment assignment and timeline for the five groups of mice. After measuring baseline periorbital and hindpaw responses, mice were given mTBI or sham injury and were administered saline or anti-CGRP mAb 2 h after the injury. The treatment was readministered on day 7 in all mice and again on day 14 in the mTBI/mAb group A. Periorbital (b) and hindpaw (c) allodynia was measured over a time course of 14 days. At 15 or 16 days after mTBI, all mice were exposed to BLS and tactile allodynia was measured over a 4 or 5 h time course. Two-way repeated measures ANOVA with Sidak’s multiple comparison test shows significantly elevated frequency of tactile responses in the mTBI/saline group compared to sham/saline mice at times indicated by a red asterisk (*). Significantly elevated frequency of tactile responses in the mTBI/mAb (group A) compared to sham/mAb mice is indicated by a green plus sign (+). Compared to saline-treated mTBI mice, frequency of response was reduced in mTBI/mAb group A and group B mice at times indicated by the green $ sign ($) and blue hashtag (#), respectively. Data are plotted as means and SEM; sham groups: n = 5 mice/group; mTBI/saline: n = 10; mTBI group A: n = 7; mTBI group B: n = 8 mice.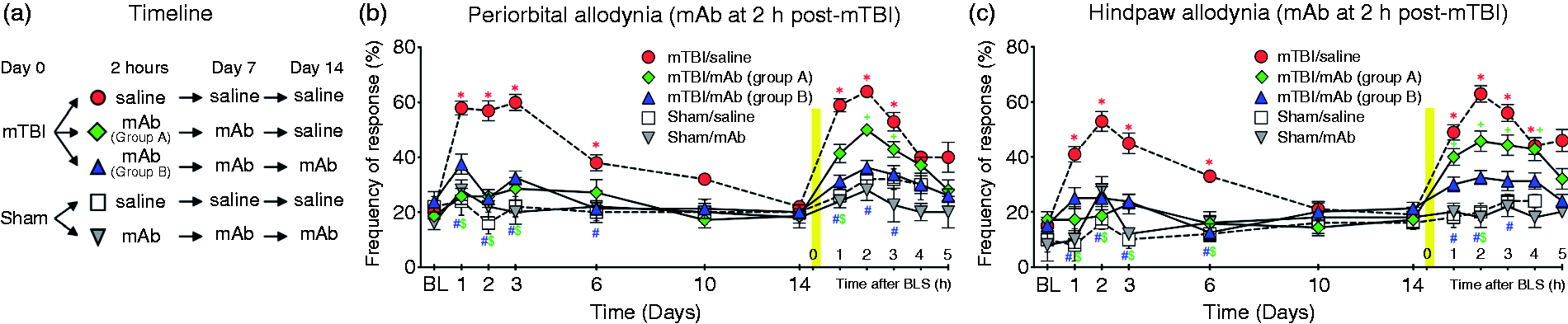
Results
mTBI increases righting time but does not produce visible injury or neurological impairment
Mice receiving mTBI under light anesthesia showed a significantly delayed righting reflex compared to sham controls (mTBI: 92.4 ± 5.4 s; n = 30 vs. sham: 35.9 ± 4.2 s; n = 20; p < 0.001, Student’s t-test), but all mice were active within 5 minutes. No skull fractures, seizures or bleeding were observed in any mice. Mice showed decreased locomotor activity in the initial post-mTBI period, though this was not quantified. No hematomas were observed with inspection of a subset of mice at necropsy.
mTBI induces immediate CA and delayed stress-induced CA
Combined data from all the mTBI (n = 39) and sham (n = 19) animals that received only control intervention (saline or control isotype antibody) are presented in Figure 1. Data from individual experiments are shown in Figures 2, 3, and in the Supplemental material, Figures S1 and S3. In mTBI mice, but not in sham mice, CA developed in both the periorbital region and hindpaw beginning on Day 1, peaking on Day 3–4 and resolving by Day 13–14. Exposure to BLS on Day 13 or 14 (depending on the experimental group) resulted in re-establishment of CA in mTBI mice but did not lead to CA in sham mice. Both periorbital and hindpaw CA peaked around the third hour after BLS exposure. Two-way ANOVA demonstrated significant effect of time, treatment group, and the interaction between the time and treatment for both periorbital and hindpaw measurements (see Table 1 for statistical data). Sidak’s multiple comparison test showed a significant difference (*p < 0.05) in CA of the periorbital region and hindpaw between mTBI and sham mice at Days 1–11 after mTBI and in post BLS hours 1–5.
The effects of anti-CGRP mAb treatment 10 days post-mTBI on tactile allodynia. (a) Periorbital and (b) hindpaw responses were measured in mice at baseline and periodically over 13 days after mTBI or sham injury. Isotype control mAb (con) or anti-CGRP mAb was administered at day 10, after the assessment of tactile responses. At day 14, all mice were exposed to BLS and tactile allodynia was measured over a 5-hour time course. Two-way repeated measures ANOVA with Sidak’s multiple comparison test shows significantly elevated frequency of tactile responses in the mTBI/con group compared to sham/con mice at times indicated by a red asterisk (*), and significantly higher responses in the mTBI/mAb group compared to sham/mAb mice indicated by a blue plus sign (+). There was no significant difference between isotype control mAb-treated and anti-CGRP mAb-treated mTBI groups at any time point during the acute and BLS-induced periorbital allodynia; hindpaw allodynia was significantly reduced only at 5 h post-BLS; blue $ sign ($). Data are plotted as means and SEM; sham: n = 5 mice/group; mTBI: n = 10 mice/group. Summary of statistical analyses.

mTBI induces CA and delayed stress-induced CA to the same degree in mice treated with saline and control antibody
In mTBI mice receiving control antibody (n = 5) or saline (n = 4), but not in the sham/control antibody mice (n = 4), CA developed in both the periorbital region and hindpaw beginning on Day 1, peaking on Day 4 and resolving by Day 14 (Figure S1). Exposure to BLS on Day 14 resulted in re-establishment of CA in mTBI mice, but not in sham mice. Two-way RM-ANOVA demonstrated significant effect of time, treatment group, and the interaction (Table 1). There was no difference between the tactile sensitivity of mTBI groups receiving saline or control antibody at any time point in either the face or hindpaw (p > 0.05) (Figure S1), and both mTBI groups exhibited significantly greater CA at days 1, 4 and 8 after mTBI and at 1–5 h time points after BLS, compared to sham (*p < 0.05; Sidak’s multiple comparison test). Our pilot experiments, using sensory thresholds as outcome measures, confirmed that repeated administration of isotype control mAb had no effect on mTBI-induced and BLS-induced allodynia (data not shown).
Half-life of anti-CGRP mAb in mice
While anti-CGRP antibodies developed for clinical use display long half-lives in humans, they have shorter half-lives in rodents due to their overall higher metabolism. The present study used a mouse anti-CGRP monoclonal antibody that shows a typical PK in mice with a half-life of 52.4 hours (Figure S2). This information was used to determine optimal dosing that would allow continuous sequestration of CGRP. Based on the half-life, three doses delivered approximately every 6 days were required to ensure continuous high levels of anti-CGRP mAb throughout the duration of the experiment.
Early, repeated anti-CGRP mAb treatment blocks immediate mTBI-induced CA
The injury assignment and drug administration time-line for five groups of mice receiving early treatment, starting 2 hours post mTBI, is shown in Figure 2(a). Compared to sham/saline mice (n = 5), mTBI mice treated with saline (n = 10) demonstrated significantly greater responsiveness to periorbital and hindpaw stimuli at Days 1–6 post mTBI returning to baseline by day 14 (*p < 0.05, Figure 2(b) and (c)). Post mTBI, Group A (n = 7) and Group B (n = 8) mice that were treated with anti-CGRP mAb at 2 hours and again at day 7 did not display a significant increase of tactile sensitivity compared to sham/mAb mice (n = 7) at any time during the 14 days after mTBI/sham (p > 0.05, Figure 2(b) and (c)). Importantly, both Groups A and B had significantly lower CA compared to mTBI/saline mice on days 1, 2, 3 and 6 (except for periorbital allodynia on day 6 in group A) (#p < 0.05, Figure 2(b) and (c)). Note that prior to day 14, Groups A and B received identical treatment and there was no difference between these groups in CA (p > 0.05).
Early, repeated anti-CGRP mAb treatment blocks subsequent stress-induced CA
On day 14, all mice received their final treatment injection (Group A and Group B mTBI mice received saline and mAb, respectively), after which they were exposed to BLS for 15 minutes. Group B mice (which received three systemic injections of anti-CGRP mAb 6 days apart over 14 days) displayed no significant stress-induced CA at any time after BLS (p > 0.05, Figure 2(b) and (c)). Group B tactile responses were similar to sham mice (saline or anti-CGRP mAb treated). By comparison, Group A mice (which received only two administrations of anti-CGRP mAb and one saline treatment), developed CA post BLS that was significantly larger than that of sham/mAb mice (*p < 0.05 at 2 and 3 hours after BLS for periorbital and at 1–4 hours for hindpaw CA), but significantly smaller than CA of mTBI/saline mice (#p < 0.05 at 1 hour after BLS for periorbital and at 2 hours for hindpaw). mTBI mice that received only vehicle treatment over the 14-day time course exhibited the strongest periorbital and extracephalic CA (*p < 0.05, compared to sham/saline, Figure 2(b) and (c)). In both periorbital and hindpaw CA responses, 2-way RM-ANOVA showed significant effects of time, treatment group, and the interaction (Table 1).
Delayed anti-CGRP mAb treatment does not prevent stress-induced CA
Post mTBI, mice (n = 10 for each treatment group) displayed significant periorbital and hindpaw CA compared to sham animals (n = 5 for each treatment group) on all testing days between Days 1–10 (*p < 0.05; Figure 3(a) and (b)). After the assessment of CA on day 10, mice received a single injection with either control antibody or anti-CGRP mAb. Mice were exposed to BLS at day 14. Regardless of treatment modality, mTBI mice developed significant stress-induced periorbital and hindpaw CA during the 5 hours post BLS (*p < 0.05, Figure 3(a) and 3(b)). There was no significant difference (p > 0.05) between isotype control antibody and anti-CGRP mAb-treated mTBI groups at any time point during the acute and BLS-induced periorbital CA or hindpaw CA except for hindpaw CA at 5 hours post BLS (#p < 0.05). CA was not observed in sham mice. Two-way RM-ANOVA demonstrated significant effect of time, treatment group, and the interaction (Table 1).
Given the half-life of the anti-CGRP mAb in mice, a separate experiment was conducted to ensure that a high blood concentration of anti-CGRP mAb was present during the exposure to BLS. To accomplish this, sham and mTBI mice were administered either isotype control antibody or anti-CGRP mAb on day 13 post sham/mTBI and were exposed to BLS 12 hours later (i.e. well within the 52.4 hours half-life of the anti-CGRP mAb and after the peak anti-CGRP mAb detection in the blood has been reached). The same outcome was observed; sham animals did not develop CA, but both mTBI groups, regardless of anti-CGRP mAb or control antibody treatment, developed significant periorbital and hindpaw allodynia at 1–4 hours after BLS (*p < 0.05, Supplemental material, Figure 3(a) and (b)). Two-way RM-ANOVA demonstrated significant effect of time, treatment group, and the interaction (Table 1).
Discussion
Summary
There are numerous notable findings in this study. First, mTBI induced by this model elicits CA for 1 to 2 weeks post injury, which can be re-instated by exposure to an environmental stressor (i.e. bright light). Re-emergent CA triggered by an external stress stimulus is particularly relevant to PPTH as stress is known to exacerbate both PTH and migraine in humans. It is important to note that the mouse mTBI model used here reproduces many mechanistic aspects of common human mild head injuries including closed-skull, blunt force direct impact, acceleration and shearing forces from unrestrained head movement. Moreover, like humans with concussion, structural damage is most often not detectable after a single mTBI by brain imaging, as well as gross and histological examination (28–31). The use of short-acting inhalation anesthetic (isoflurane) is specific to the animal model and is noted as a limitation.
Second, both cephalic and extracephalic CA were consistently induced by mTBI and subsequently by BLS. The presence of cephalic and extracephalic CA suggests that mTBI is likely to promote sensitization of both trigeminal pathways as well as brain pain modulatory pathways that may promote generalized allodynia and that both of these processes begin soon after injury.
Third, treatment with anti-CGRP mAb, starting early (2 hours) after mTBI and repeated twice more throughout the 14-day time course to promote sequestration of CGRP neuropeptide, reduced the cephalic and extracephalic tactile sensitivity to levels comparable to those of sham animals both immediately after mTBI as well as after exposure to BLS. In contrast, repeated administration of saline or isotype control antibody had no effect on CA in mTBI mice. This suggests that mTBI-induced CA and establishment of a sensitized state are CGRP-dependent mechanisms.
Fourth, when a delayed, single injection of anti-CGRP mAb was given after CA had begun to resolve (day 10), it failed to prevent stress-induced CA in mice that had received an mTBI. These data suggest that following establishment of a sensitized state, additional CGRP-independent mechanisms promote or maintain central sensitization and anti-CGRP mAb treatment is not effective. It should be noted that the time dependency of establishment of sensitized states in humans is not known and likely differs from the timeline determined in the current mouse study.
Finally, when anti-CGRP mAb was given only at 2 hours and 7 days and not again at 14 days, the suppression of initial mTBI-induced CA out to 14 days occurred, but only a partial reduction in BLS-induced CA was observed. This raises the possibility that CGRP signaling continues after the resolution of initial CA and contributes to the maintenance of central sensitization and to vulnerability to provocative stimuli that may produce PPTH. Therefore, anti-CGRP mAb treatment starting early after mTBI and continuing beyond the resolution of acute CA is likely required for full therapeutic benefits.
Animal model of PTH and PPTH
The mouse model of mTBI used in this study reproduces many mechanistic aspects of common human head injuries. Closed-head injury models largely recapitulate most human mild head injuries, and in the animal model, they eliminate additional and confounding sensitization that is likely to occur with other TBI models that breach the scalp and calvarium. This closed head injury model delivers a high velocity direct impact which is analogous to the most common mechanism of human mTBIs. The head is not fixed and able to move freely upon impact. This allows for the generation of both linear acceleration and rotational acceleration, common features of impact injuries that contribute to and may be necessary for producing pain and the behavioral sequelae that occur in concussion (33–35). Therefore, this model of mTBI appears particularly appropriate for investigating mechanisms and interventions relevant to human concussion.
We used a variation of the “two-hit” strategy of hyperalgesic priming (36) that has been recently employed in pain research, whereby an initial noxious stimulus confers a significant response to a second subthreshold stimulus delivered following resolution of pain from the initial injury. The interpretation of such observations from many different laboratories is that the initial insult lays down a neural memory of pain that results in amplification of inputs in central pathways and that may reflect mechanisms that underlie transition from episodic to chronic pain states (37). In our studies, the mTBI was literally the “first hit,” revealing the consequences of a provocative, second hit stimulus induced by a normally innocuous exposure to stress from bright light (BLS). Both bright light and stress are frequently reported aggravating factors of both migraine and PTH (18,38). Photophobia was reported more frequently in PTH patients one year after concussion than in patients one year after traumatic orthopedic (non-head) injury, despite comparable rates of headache and migraine (39) in the two groups. Our studies suggest that mTBI produces long-lasting vulnerability to pain from provocative stimuli following establishment of central sensitization.
PTH and cutaneous allodynia
Post-traumatic headache is the most frequently reported symptom of concussion, and it often has a migraine-like phenotype (1). As it is a challenge to determine if animals are experiencing ongoing pain or headache, behavioral/stimulus-induced measures are often employed as a surrogate for pain and headache. In particular, allodynia, pain in response to non-painful stimuli, is often measured in animal models. CA is common during a migraine attack (40–42) and is also reported in over 50% of patients with post-traumatic headache (43,44). When sensory thresholds were tested in human subjects, 53% of patients with PTH displayed cephalic allodynia compared to only 20% of TBI patients who did not have PTH and 0% of control subjects (44). It is especially notable that in the mTBI mouse model used in this study, allodynia was observed in both cephalic and hindpaw regions. Patients with migraine also often experience both cephalic and extracephalic allodynia (45).
Previous preclinical studies examining the mechanisms underlying allodynia after TBI have suggested the occurrence of activation and sensitization of afferent fibers from the dura mater, the scalp and skin as well as sensitization of second order neurons in the trigeminal nucleus caudalis (TNC) where these peripheral afferents converge (46,47). As nociceptive pathways for pain sensation in the rodent hindpaw and human forearm do not involve the TNC, sensitization of higher-order circuits in the pain pathway is also likely to occur. Preclinical studies modeling migraine have suggested that sensitization of thalamic neurons may be responsible for development of extracephalic allodynia (46,48).
CGRP and migraine
CGRP is a potent vasodilator throughout the body, including the cerebral vasculature. However, the role of CGRP in headache pain is not thought to be dependent on its vasodilatory effects but rather on its involvement in neurotransmission in the trigeminal sensory system. While not fully established, this is thought to take place predominantly at the peripheral afferent neurons (49), in part due to the efficacy of the anti-CGRP mAbs, which do not significantly cross the blood brain barrier. Nonetheless, CGRP and CGRP receptors have been identified in the human and animal brain and brainstem, including many areas that are involved in nociception and pain processing (50–52).
CGRP has been shown to play a causal role in the generation of migraine (see Edvinsson (22) for review). Briefly, CGRP and CGRP receptors are present in over half of the trigeminal nerve fibers and neurons in both the central and peripheral nervous system (49,50,53). In a study examining patients with neuralgia and also using a cat animal model, stimulation along the trigeminal pathway resulted in increases in CGRP blood levels (54). CGRP administered intravenously to migraine patients evokes attacks that are highly similar to spontaneous migraine attacks (55). There is significant CGRP release in the ictal stage of acute migraine (56). CGRP levels predict response to triptans (57) and reduce simultaneously with effective treatment of migraine with triptans (58). Patients with migraine have higher concentrations of CGRP in the blood than controls and CGRP is chronically elevated in patients with chronic migraine (59). Triptans, migraine specific medications used in the acute setting to halt the migraine process, have presynaptic receptors that inhibit the release of CGRP and post-synaptic receptors that counteract CGRP vasodilation (60).
These observations have led to the development of therapies for the treatment and prevention of migraine that target the CGRP pathway. These therapies include monoclonal antibodies against CGRP (61–63) and the CGRP receptor (64) as well as CGRP antagonists (the gepants) (65). The monoclonal antibodies have been shown in clinical trials to be effective for the prevention of migraine (61–64) and have been prescribed to over 100,000 patients since their release in May 2018 (66). Anti-CGRP antibodies have also been recently demonstrated to be effective in the treatment of cluster headache (67,68). Additionally, the gepants are currently being investigated for their safety and efficacy as abortive and preventive therapies for migraine (69–71).
CGRP and PTH
In addition to its role as a potent vasodilator and neurotransmitter in trigeminal nociception, CGRP has been implicated to play a role in inflammatory pain, with up-regulation of CGRP and CGRP mRNA in the DRG observed in many induced and chronic inflammatory pain states (see Iyengar et al. (72) for review]. Additionally, perivascular release of CGRP from the trigeminal afferent nerves generates vasodilation and mast cell degranulation (73,74). Both vasodilation and mast cell degranulation play a role in neurogenic inflammation, and mast cells have been implicated in the pathophysiology of migraine (75). CGRP receptors are also found on the satellite glial cells of the TG. Thus, CGRP may be particularly relevant to PTH as traumatic brain injury initiates a cascade of events, including an inflammatory response with activation of mast cells, microglia and satellite cells, release of cytokines, chemokines and other inflammatory modulators and alterations in the blood brain barrier (see Mayer et al. (76) for review).
In a controlled cortical impact (CCI) mouse model of TBI, CCI was associated with increased periorbital cutaneous allodynia that correlated with increases in CGRP in the brainstem. CCI also increased the number of microglia in the injured cortex. However, a decline in the number of these cells over the weeks post injury suggested that the persistent allodynia and increased CGRP and other inflammatory mediators were maintained by mechanisms other than activation of microglia (77). Additionally, in studies using a blast-injury mouse model or weight-drop model in mice or rats for mTBI, mast cell degranulation was found to persist for up to 30 days post injury (78,79).
Using a fixed head weight-drop method in rats, Bree and Levy (24) reported the development of periorbital, but not hindpaw, CA that was resolved within the first 14 days after mTBI (24). Moreover, pain could be re-evoked after return to baseline by administration of glyceryl trinitrite (GTN), a compound well known to trigger migraine in humans (24). Consistent with our results, administration of mouse anti-CGRP mAb every 6 days prevented both periorbital allodynia and GTN-evoked re-emergence of periorbital CA at days 15 and 30 post TBI. Collectively, these studies confirm that sequestering CGRP after mTBI prevents both CA and responses to migraine triggers in rodents.
Some differences between the two studies are notable, however. Unlike our study, the previous work used a weight drop method with a fixed head (24), preventing linear and rotational forces that are commonly associated with closed head impact injuries. Using these methods, cephalic, but not extra-cephalic, allodynia was observed, leading the authors to conclude that PTH-related pain may result from sensitization of peripheral nociceptors without central sensitization (24). In contrast, our study allowing linear and rotational head movement upon weight drop, as well as a previous report (80) that allowed for anteroposterior motion without any rotational movement, showed both cephalic and extra-cephalic allodynia in mice exposed to mTBI, suggestive of centrally sensitized states. Differences in observed results could reflect experimental procedures and species studied. While our data and a previous study in rats (24) both showed that continuous CGRP sequestration prevented the pain states that were reinstated by triggers including GTN or stress, we additionally demonstrated that delayed administration of anti-CGRP mAb was not effective in blocking stress-provoked pain behaviors. Thus, although CGRP appears critical in establishing a sensitized state that promotes vulnerability to headache triggers, the evoked pain-like response that may represent PPTH is not dependent exclusively upon CGRP. It remains possible that early intervention after mTBI could show efficacy due to impaired integrity of the blood brain barrier allowing for central actions of anti-CGRP mAbs; this possibility requires further investigation. Alternatively, repeated administration of anti-CGRP mAb following established mTBI-induced sensitization could be effective in ameliorating sensitivity to triggering events. In regard to possible non-CGRP mechanisms, we note that while CGRP is a clinically validated mediator of migraine, not all patients respond to indirect modulators of CGRP such as triptans (81). Similarly, not all migraine patients respond to anti-CGRP mAbs, and in the majority of responders, even though a reduction in attacks is experienced, breakthrough attacks still occur (61–64,82). Especially relevant is that, in general, the response to anti-CGRP mAbs is less robust in patients with chronic migraine compared to patients with episodic migraine (82). The experience of continuing migraine attacks in patients on anti-CGRP mAb therapy suggests CGRP-independent mechanisms.
Our findings suggest that CGRP-dependent mechanisms are involved in the development of central sensitization after mTBI and, by translation, the transition from acute and episodic to chronic pain states, including PPTH. A CGRP ligand targeted mAb in these studies prevented this transition but was ineffective when administered after the establishment of sensitized state. Whether anti-CGRP mAbs will be effective clinically in treating acute or persistent PTH, or prevent the development of PPTH, remains to be established. Important questions include whether a similar window of opportunity exists in humans, the duration of this window, and whether repeated concussions, repetitive sub-concussive head impacts, or a previous history of migraine may be risk factors in promoting PPTH and resistance to CGRP pathway targeted therapies. We note that a limitation of our study was the use only of male mice and that sex is an established biological variable, with females showing increased risk for PTH following mTBI (83).
Conclusion
The results presented in this study contribute to the understanding of the mechanism of APTH and PPTH and the role of CGRP in these processes. The data support a CGRP-dependent mechanism that initiates a sensitized state that likely underlies both APTH and PPTH. Additionally, our data indicate that once central sensitization is established, CGRP is not directly responsible for the pain associated with stress, a common migraine and PTH-related trigger suggesting CGRP-independent mechanisms. Furthermore, these data provide insights into the possible use and timing of anti-CGRP mAb for patients with mTBI for the treatment of APTH and the prevention of emergence of PPTH.
Article highlights
A mouse model of mild traumatic brain injury (mTBI) was adapted to evaluate novel pain-related measures of acute (APTH) and persistent (PPTH) post-traumatic headache. mTBI produced generalized periorbital and hindpaw cutaneous allodynia suggestive of PTH-related pain and central sensitization. Following resolution of mTBI-induced cutaneous allodynia, stress reinstated CA only in previously injured mice, suggestive of sustained central sensitization and vulnerability to PPTH. Continuous sequestration of CGRP prevented mTBI-related APTH as well as stress-induced PPTH. A single administration of anti-CGRP mAb after establishment of central sensitization was not effective in blocking PPTH-related pain, suggesting CGRP-independent mechanisms. Early and sustained targeted intervention following mTBI with anti-CGRP antibodies treatment may prevent debilitating PTH and its persistence.
Supplemental Material
CEP877662 Supplemental Material - Supplemental material for CGRP-dependent and independent mechanisms of acute and persistent post-traumatic headache following mild traumatic brain injury in mice
Supplemental material, CEP877662 Supplemental Material for CGRP-dependent and independent mechanisms of acute and persistent post-traumatic headache following mild traumatic brain injury in mice by Edita Navratilova, Jill Rau, Janice Oyarzo, Jason Tien, Kimberly Mackenzie, Jennifer Stratton, Bethany Remeniuk, Todd Schwedt, Trent Anderson, David Dodick and Frank Porreca in Cephalalgia
Footnotes
Acknowledgements
EN, TA and FP conceived and planned the study; JO, JT and KM performed experiments; EN, JT and KM analyzed data; FP, BR, JR and EN wrote the manuscript; EN, JR, TA, JS, KM, JT, TS, DD and FP corrected and improved the manuscript. TA, JS, TS, and DD consulted on methods and clinical relevancy and proof-read the manuscript.
Declaration of conflicting interests
The authors declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: TJS has consulted for Alder, Allergan, Amgen, ATI, Avanir, Cipla, Dr. Reddy’s, Eli Lilly, Ipsen Bioscience, Nocira, Novartix, Teva, and Xoc. He holds stock options in Aural Analytics, Nocira, and Second Opinion. He receives research funding from Amgen. JS, JT and KM are employees of TEVA Pharmaceuticals. FP has received research funding from Voyager, Hoba, Allergan, PeptideLogic, Proximagen, Ipsen and Eli Lilly. DWD reports the following conflicts: Personal fees: Amgen, Association of Translational Medicine, University Health Network, Daniel Edelman Inc., Autonomic technologies, Axsome, Aural Analytics, Allergan, Alder, Biohaven, Charleston Laboratories, Dr Reddy’s Laboratories/Promius, Electrocore LLC, Eli Lilly, eNeura, Neurolief, Novartis, Ipsen, Impel, Satsuma, Supernus, Sun Pharma (India), Theranica, Teva, Vedanta, WL Gore, Nocira, PSL Group Services, University of British Columbia, XoC, Zosano, ZP Opco, Foresite Capital, Oppenheimer. CME fees or royalty payments from Healthlogix, Medicom Worldwide, Medlogix Communications, Mednet, Miller Medical, PeerView, WebMD Health/Medscape, Chameleon, Academy for Continued Healthcare Learning, Universal meeting management, Haymarket, Global Scientific Communications, Global Life Sciences, Global Access Meetings, UpToDate (Elsevier), Oxford University Press, Cambridge University Press, Wolters Kluwer Health; Stock options: Aural analytics, Healint, Theranica, Second Opinon/Mobile Health, Epien, GBS/Nocira, Matterhorn/Ontologics, King-Devick Technologies. Consulting without fee: Aural Analytics, Healint, Second Opinion/Mobile Health, Epien; Board of Directors: Epien, Matterhorn/Ontologics, King-Devick Technologies. Patent: 17189376.1-1466: Title: Botulinum Toxin Dosage Regimen for Chronic Migraine Prophylaxis without fee. Professional society fees or reimbursement for travel: American Academy of Neurology, American Brain Foundation, American Headache Society, American Migraine Foundation, International Headache Society, Canadian Headache Society. Other: Use agreement through employer: Myndshft.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by an unrestricted grant from TEVA to FP.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.