
Abstract
Objective
The objective of this study was the determination of the role of calcitonin gene-related peptide (CGRP) in the induction of medication overuse headache (MOH)-related migraine in an injury-free preclinical model.
Methods
Rats were primed by a 7-day period of exposure to acute migraine therapies including sumatriptan and morphine. After an additional 14-day drug-free period, rats were exposed to putative migraine triggers including bright light stress (BLS) or nitric oxide (NO) donor in the presence or absence of TEV48125, a fully humanized CGRP antibody. Cutaneous allodynia (CA) was used as an outcome measure and CGRP blood and cerebrospinal fluid (CSF) levels were measured.
Results
BLS and NO donor challenge evoked delayed, long-lasting CA selectively in rats that were previously treated with sumatriptan or morphine. BLS produced a significant increase in CGRP in the plasma, but not CSF, in animals that were previously exposed to sumatriptan compared to saline controls. TEV48125 did not modify baseline tactile thresholds or produce behavioral side effects, but significantly inhibited both BLS- and NO donor-induced CA in animals that were previously primed with sumatriptan or morphine; an isotype control protein that does not bind CGRP had no effect.
Interpretation
These data suggest that acute migraine medications may promote MOH in susceptible individuals through CGRP-dependent mechanisms and that anti-CGRP antibodies may be a useful clinical strategy for the treatment of MOH.
Introduction
Migraine is the world’s most common neurological disorder and greatly diminishes quality of life (1). Attacks are characterized by episodic, intense headaches that are unilateral and throbbing and usually accompanied by nausea, photophobia or phonophobia and may occur with or without aura (1). Repeated or excessive use of acute migraine therapies in predisposed patients can lead to medication overuse headache (MOH) (2). MOH is characterized by 15 or more headache days per month for at least 3 months. The threshold number that constitutes excessive use of acute migraine therapies is defined by the medication being overused typically being more than 10–15 days per month (2). Development of MOH is influenced by the type of medication, magnitude of exposure (which varies as a function of medication class) and the gender and age of the individual. Opioids and triptans, as well as compounds with barbiturates and ergotamine, have significant potential for inducing MOH (2). The mechanisms involved in the development and maintenance of MOH remain uncertain.
We developed a rodent model of MOH based on sustained triptan or morphine exposure that induces a long-lasting state of “latent sensitization” (3,4). Systemic exposure of rats to triptans or opiates over a period of 1 week produces cutaneous allodynia (CA) during the period of drug administration that resolves within 2 weeks following termination of the drug. Challenge of the triptan- or morphine-exposed rats with presumed migraine triggers including environmental stress (exposure to a bright light) or a nitric oxide (NO) donor produces delayed, long-lasting and generalized (i.e., periorbital and hind-paw) CA that is accompanied by increased expression of c-FOS in the trigeminal nucleus caudalis (TNC), suggesting activation of the trigeminal nociceptive pathways (3,4).
Calcitonin gene-related peptide (CGRP) is understood to be important in the pathophysiology of migraine (3,5). Release of CGRP from nociceptors innervating the cranial meninges contributes to neurogenic vasodilation and the release of mediators that can result in the sensitization and activation of these fibers (6). Sustained activation of nociceptors can elicit sensitization of central pain processing pathways, resulting in migraine pain (7). Many migraine patients experience CA in the ipsilateral cephalic region that can spread contralaterally and to the upper torso and limbs (7). Generalized CA likely reflects central sensitization and has been a useful translational endpoint for preclinical studies (3). The role of CGRP in CA is supported by the significantly increased levels of this peptide in the plasma during migraine attacks, as well as during interictal periods, in patients with chronic migraine (8,9). Additionally, provocative stimulation with CGRP or NO donors evokes migraine in migraineurs, but not in non-migraineurs (8,10). We wished to determine whether CA resulting from common migraine triggers in a rat model of MOH was dependent upon CGRP. We used TEV48125, a fully humanized CGRP antibody that is effective in the preventative treatment of episodic and chronic migraine (11).
Materials and methods
Animals
Adult male Sprague–Dawley rats (Harlan Laboratories, Inc., Indianapolis, IN, USA), initially weighing 175–225 g, were maintained three per cage in a climate-controlled room at 22 ± 2℃ on a 12-hour light/dark cycle (lights on at 7 am–7 pm) with free access to food and water. Studies were conducted during the animals’ light cycle following approval by the Institutional Animal Care and Use Committee of the University of Arizona.
Drugs
Sumatriptan succinate (Abmole Bioscience, Houston, TX, USA) and sodium nitroprusside (SNP; Sigma-Aldrich, St. Loius, MO, USA) were dissolved in sterile 0.9% saline solution. Morphine (75 mg each) or placebo pellets were generously provided by the National Institutes on Drug Abuse. TEV48125 and an isotype control antibody were provided by Teva Biologics (Redwood City, CA, USA) and reconstituted in saline just before use.
General experimental design overview
We assessed the influence of TEV48125 (a CGRP monoclonal antibody), a control isotype antibody and vehicle (saline) on periorbital and hind-paw tactile thresholds following exposure to two presumed migraine triggers (i.e., NO donor injection or two sessions of environmental (i.e., bright light) stress). The putative migraine triggers that were used have been previously described (3,12). All studies were performed by experimenters (CMK and NME) who were blinded to the treatment conditions, with a different person randomly assigning each animal to different groups and dosing the animals.
Induction of sumatriptan or morphine latent sensitization models of MOH
Following transient anesthesia (2% isoflurane), the animals received either subcutaneous 7-day osmotic mini-pumps (Model 2001, Alzet, Cupertino, CA, USA) containing sumatriptan (0.6 mg/kg/day) or saline, or two morphine (75 mg each) or placebo pellets to produce sustained sumatriptan or morphine exposure over 7 days, respectively (4). These compounds were given over a period of 1 week, and no experiments were performed for an additional 2 weeks allowing washout of the drugs, eliminating potential concerns about differences in pharmacokinetics.
Evaluation of CA
Animals were placed in individual chambers in a quiet environment and allowed to acclimatize for 45–60 minutes (13). Withdrawal thresholds to tactile stimulation of the periorbital region or the hind-paw were determined using von Frey filaments, as previously described (3). Baseline withdrawal thresholds were measured prior to drug infusion and again on day 21 prior to challenge with provocative migraine stimuli (3).
Antibody treatment
On day 18 post-pellet or mini-pump implantation, rats received either a single injection of TEV48125 (10 or 30 mg/kg subcutaneously), its vehicle (saline) or the isotype control protein (30 mg/kg subcutaneously) at 1 ml/kg.
Provocative challenge
Rats were divided into two cohorts and challenged with provocative migraine triggers including bright light stress (BLS) or a NO donor in order to determine the possible influence of TEV48125 on periorbital and hind-paw tactile thresholds.
Rats received two episodes of BLS on days 20 and 21 post-drug implantation. BLS was induced by two halogen lights that were placed on both sides of the home cage to deliver between 3400 and 3700 lux for 1 hour each day. Tactile thresholds were measured prior to BLS and at 1-hour intervals after the second BLS on day 21 for 5 hours (12).
Separate groups of rats received SNP (3 mg/kg intraperitoneally), a NO donor. Periorbital and hind-paw tactile thresholds were measured similarly before and after SNP administration (3).
Measurement of CGRP levels
Concentrations of CGRP in plasma and cerebrospinal fluid (CSF) were measured with commercially purchased enzyme-linked immunosorbent assay kits (Cayman Chemical, Ann Harbor, MI, USA). Plasma and CSF were collected from the same animals following BLS given on days 20 and 21. At 0.5 or 2 hours after the second BLS on day 21, separate groups of animals were anesthetized with ketamine/xylazine (80/12 mg/kg intraperitoneally; Western Medical Supply, Arcadia, CA, USA; Sigma-Aldrich, St. Louis, MO, USA). Whole blood (3 ml) was extracted from the jugular vein and stored in ethylenediaminetetraacetic acid-coated vials. The animals were fixed onto a stereotaxic frame and the atlanto-occipital membrane was exposed surgically to enable CSF extraction. Collection of both blood and CSF took approximately 5 minutes per animal. Blood was centrifuged at 3000 rpm for 20 minutes at 4℃ in order to separate the plasma. Both plasma and CSF samples were stored at −80℃ and analyzed within 1 week of collection (3).
Statistical analysis
Two-way analysis of variance (ANOVA) followed by Dunnett’s multiple comparisons test were used for comparisons of treatment and time effects. Area over the time–effect curves (AOCs) were calculated using Flashcalc created by Dr. Michael Ossipov (http://www.u.arizona.edu/∼michaelo/). Between-group differences were compared using one-way ANOVA post hoc Tukey’s multiple comparison tests for the AOC data and unpaired t-tests (two-tailed) for CGRP concentrations. Statistical significance was set at p < 0.05. All data are presented as mean ± SEM. Outliers were identified by Grout tests and were excluded from the analysis.
Results
Dose-dependent blockade of NO donor-induced CA by TEV48125 in rats with sumatriptan-induced latent sensitization
No differences in baseline sensory thresholds were observed in rats that had previously received saline or sumatriptan on day 21 after mini-pump implantation. SNP evoked delayed and long-lasting periorbital and hind-paw CA selectively in rats with prior sumatriptan infusion (Figure 1). Significant allodynia at periorbital (Figure 1(a)) and hind-paw (Figure 1(c)) regions was detected at 2, 3 and 4 hours and at 1, 2, 3 and 4 hours post-SNP administration, respectively. No significant change of tactile threshold was observed in rats that were pretreated with saline.
Blockade of cutaneous allodynia by systemic administration of TEV48125 evoked by nitric oxide (NO) donor injection in rats with sumatriptan-induced latent sensitization. On day 18 after the initial exposure of sumatriptan (0.6 mg/kg/day for 7 days subcutaneously) or its vehicle (saline, 1 ml/kg/day), rats received TEV48125 at 10 or 30 mg/kg subcutaneously or its vehicle (saline, 1 ml/kg subcutaneously). At day 21 post-sumatriptan exposure, periorbital and hind-paw tactile thresholds were evaluated prior to baseline (BL) and hourly after NO donor injection (3 mg/kg intraperitoneally) for up to 5 hours. (a) and (b) represent periorbital tactile threshold time courses and area over the time–effect curves (AOCs), respectively. (c) and (d) represent hind-paw tactile threshold time courses and AOCs, respectively. Values are means ± SEM of 13–20 rats per group. In (a) and (c), * indicates p < 0.05 compared to pre-dose baseline. In (b) and (d), * and #indicate p < 0.05 sumatriptan/vehicle versus saline/vehicle or sumatriptan/TEV48125 (30 mg/kg), respectively. Two-way analysis of variance (ANOVA) post hoc Dunnett’s multiple comparisons tests were applied for the tactile threshold data. One-way ANOVA post hoc Tukey’s multiple comparison tests were used in order to analyze the AOC data.
Systemic treatment with TEV48125 (10 and 30 mg/kg subcutaneously) on day 18 after mini-pump implantation did not change either baseline thresholds on day 21 prior to NO donor (Figure 1(a) and 1(c)). TEV48125 (10 mg/kg) partially reduced NO donor-induced CA. Significant hind-paw allodynia was only observed at 2 hours post-SNP in the sumatriptan/TEV48125 10 mg/kg group (p < 0.01 compared to day 21 baseline). A higher dose of TEV48125 (30 mg/kg subcutaneously) resulted in complete prevention of NO donor-induced CA in rats that were pre-exposed to sumatriptan (Figure 1; p > 0.05 at any time point). There was a significant difference between the sumatriptan/TEV48125 (30 mg/kg) and sumatriptan/vehicle rats at 2 hours (p < 0.05) post-BLS for hind-paw CA. It is noteworthy that TEV48125 did not change the tactile threshold of rats that were chronically infused with saline.
The integrated AOC calculated from day 21 baseline to 5 hours post-NO donor injection demonstrated a significant dose-dependent inhibition of CA (Figure 1(b) and 1(d)) by TEV48125 in rats that were primed with sumatriptan (p < 0.01 sumatriptan/vehicle vs. sumatriptan/TEV48125 30 mg/kg or saline/vehicle group). In order to control for the potential effects on pain behaviors that may be caused by high doses of antibody treatment, we evaluated the effect of an isotype control antibody containing the same Fc format as TEV48125 on NO donor-induced CA in separate groups of rats that had previously received sumatriptan or saline as described above. Animals were treated at day 18 with the isotype control protein (30 mg/kg subcutaneously) or vehicle and challenged with SNP (3 mg/kg intraperitoneally) at day 21 post-sumatriptan infusion. No difference in NO donor-induced CA was detected between the sumatriptan/vehicle and sumatriptan/control protein groups. The AOC of periorbital allodynia for the sumatriptan/control protein group was not significantly different from that of the sumatriptan/vehicle group (1.9 ± 0.5 and 2.3 ± 0.9, p > 0.05). Similar results were obtained for the AOC hind-paw allodynia from the sumatriptan/control protein and sumatriptan/vehicle groups; the AOCs were 10.0 ± 4.1 and 9.8 ± 4.0 (p > 0.05), respectively. No CA was observed in animals that were pretreated with saline and challenged with the NO donor and no effects of the isotype control protein were observed in these groups (data not shown).
TEV48125 abolished BLS-induced CA after sumatriptan-induced latent sensitization
Exposure to two episodes of BLS on consecutive days produced long-lasting periorbital and hind-paw CA selectively in rats that were primed with sumatriptan (Figure 2). Significant CA (Figure 2(a) and 2(c)) was detected at 2, 3 and 4 hours and at 2, 3, 4 and 5 hours post-stress, respectively, in the sumatriptan/vehicle group (p < 0.05 vs. day 21 baseline). No significant changes in tactile thresholds were observed in the saline/vehicle group (p > 0.05 for all time points compared to pre-BLS baseline). The possible effect of CGRP blockade on BLS-induced allodynia was evaluated by a single pretreatment dose of TEV48125 (30 mg/kg) on day 18. TEV48125 prevented BLS-induced CA in rats that were pre-exposed to sumatriptan (Figure 2; p > 0.05 at any time point vs. day 21 baseline). Rats that were previously exposed to saline did not develop tactile allodynia and TEV48125 did not alter sensory thresholds in these animals. The integrated AOC of each treatment calculated from day 21 baseline to 5 hours post-stress also showed that TEV48125 produced significant blockade of BLS-induced CA (Figure 2(b) and 2(d); p < 0.01 sumatriptan/vehicle vs. sumatriptan/TEV48125 30 mg/kg or saline/vehicle group).
TEV48125 blocked cutaneous allodynia evoked by bright light stress (BLS) in rats with sumatriptan-induced latent sensitization. TEV48125 (30 mg/kg subcutaneously) or its vehicle (saline, 1 ml/kg subcutaneously) were injected at day 18 post-sumatriptan (0.6 mg/kg/day for 7 days subcutaneously) or saline (1 ml/kg/day subcutaneously) infusion. Rats were exposed to BLS for 1 hour on days 20 and 21 after sumatriptan administration. At day 21, periorbital and hind-paw tactile thresholds were evaluated prior to baseline (BL), and hourly after BLS for up to 5 hours. (a) and (b) represent periorbital tactile threshold time courses and area over the time–effect curves (AOCs), respectively. (c) and (d) represent hind-paw tactile threshold time courses and AOCs, respectively. Values are means ± SEM of 21–35 rats per group. In (a) and (c), * indicates p < 0.05 compared to pre-dose baseline. In (b) and (d), * and # indicate p < 0.05 sumatriptan/vehicle versus saline/vehicle or sumatriptan/TEV48125 (30 mg/kg), respectively. Two-way analysis of variance (ANOVA) post hoc Dunnett’s multiple comparisons tests were applied for the tactile threshold data. One-way ANOVA post hoc Tukey’s multiple comparison tests were used in order to analyze the AOC data.
Blockade of NO donor-induced CA by TEV48125 in rats with morphine-induced latent sensitization
SNP evoked delayed and long-lasting periorbital and hind-paw allodynia selectively in rats with prior morphine administration (Figure 3). Significant CA (Figure 3(a) and 3(c)) was detected at 1, 2, 3 and 4 hours and at 2, 3 and 4 hours post-SNP administration, respectively (p < 0.05 vs. day 21 baseline). No significant changes of tactile threshold were observed following SNP in rats that were pretreated with placebo.
Blockade of cutaneous allodynia by systemic administration of TEV48125 evoked by nitric oxide (NO) donor injection in rats with sustained morphine-induced latent sensitization. On day 18 after the initial implantation of morphine (2 × 75 mg subcutaneously) or placebo pellets, rats received TEV48125 at 30 mg/kg subcutaneously or its vehicle (saline, 1 ml/kg subcutaneously). At day 21 post-sumatriptan exposure, periorbital and hind-paw tactile thresholds were evaluated prior to baseline (BL) and hourly after NO donor injection (sodium nitroprusside; 3 mg/kg intraperitoneally) for up to 5 hours. (a) and (b) represent periorbital tactile threshold time courses and area over the time–effect curves (AOCs), respectively. (c) and (d) represent hind-paw tactile threshold time courses and AOCs, respectively. Values are means ± SEM of 11–24 rats per group. In (a) and (c), * indicates p < 0.05 compared to pre-dose BL. In (b) and (d), * indicates p < 0.05 morphine/vehicle versus placebo/vehicle. Two-way analysis of variance (ANOVA) post hoc Dunnett’s multiple comparisons tests were applied for the tactile threshold data. One-way ANOVA post hoc Tukey’s multiple comparison tests were used in order to analyze the AOC data.
TEV48125 (30 mg/kg subcutaneously) given at day 18 post-pellet implantation did not change either periorbital or hind-paw baseline tactile thresholds assessed on day 21 prior to NO donor injection. The expression of CA induced by NO donor injection in rats that were pre-exposed to morphine was significantly diminished by TEV48125 (p > 0.05 vs. day 21 baseline at any time point). No effect of the NO donor was observed in rats that were treated with placebo pellets (p > 0.05 vs. day 21 baseline). Similar results are shown in the AOC data calculated from day 21 baseline to 5 hours post-stress (Figure 3(b) and 3(d); p < 0.01 morphine/vehicle vs. morphine/TEV48125 30 mg/kg or placebo/vehicle group).
TEV48125 abolished the BLS-induced CA after morphine-induced latent sensitization
BLS produced long-lasting periorbital and hind-paw CA selectively in rats that were treated previously with morphine (Figure 4). Significant CA (Figure 4(a) and 4(c)) was detected at 2 hours and at 2, 3 and 4 hours post-stress, respectively, in the morphine/vehicle group (p < 0.05 vs. day 21 baseline), which was significantly blocked by TEV48125 (30 mg/kg) given at day 18 (p > 0.05 vs. day 21 baseline at any time point). Rats that were previously exposed to placebo did not develop CA post-BLS and TEV48125 did not alter sensory thresholds in these animals. The integrated AOC of each treatment calculated from day 21 baseline to 5 hours post-stress also showed that TEV48125 produced a significant blockade of the development of BLS-induced CA (Figure 4(b) and 4(d); p < 0.05 morphine/vehicle vs. morphine/TEV48125 30 mg/kg or placebo/vehicle group).
TEV48125 blocked cutaneous allodynia evoked by bright light stress (BLS) in rats with sustained morphine-induced latent sensitization. TEV48125 (30 mg/kg subcutaneously) or its vehicle (saline, 1 ml/kg subcutaneously) were injected at day 18 post-implantation of morphine (2 × 75 mg subcutaneously) or placebo pellets. Rats were exposed to BLS for 1 hour on days 20 and 21 after morphine administration. At day 21, periorbital and hind-paw tactile thresholds were evaluated prior baseline (BL) and hourly after BLS for up to 5 hours. (a) and (b) represent periorbital tactile threshold time courses and area over the time–effect curves (AOCs), respectively. (c) and (d) represent hind-paw tactile threshold time courses and AOCs, respectively. Values are means ± SEM of 8–13 rats per group. In (a) and (c), * indicates p < 0.05 compared to pre-dose BL. In (b) and (d), * and # indicate p < 0.05 morphine/vehicle versus saline/vehicle or morphine/TEV48125 (30 mg/kg), respectively. Two-way analysis of variance (ANOVA) post hoc Dunnett’s multiple comparisons tests were applied for the tactile threshold data. One-way ANOVA post hoc Tukey’s multiple comparison tests were used in order to analyze the AOC data.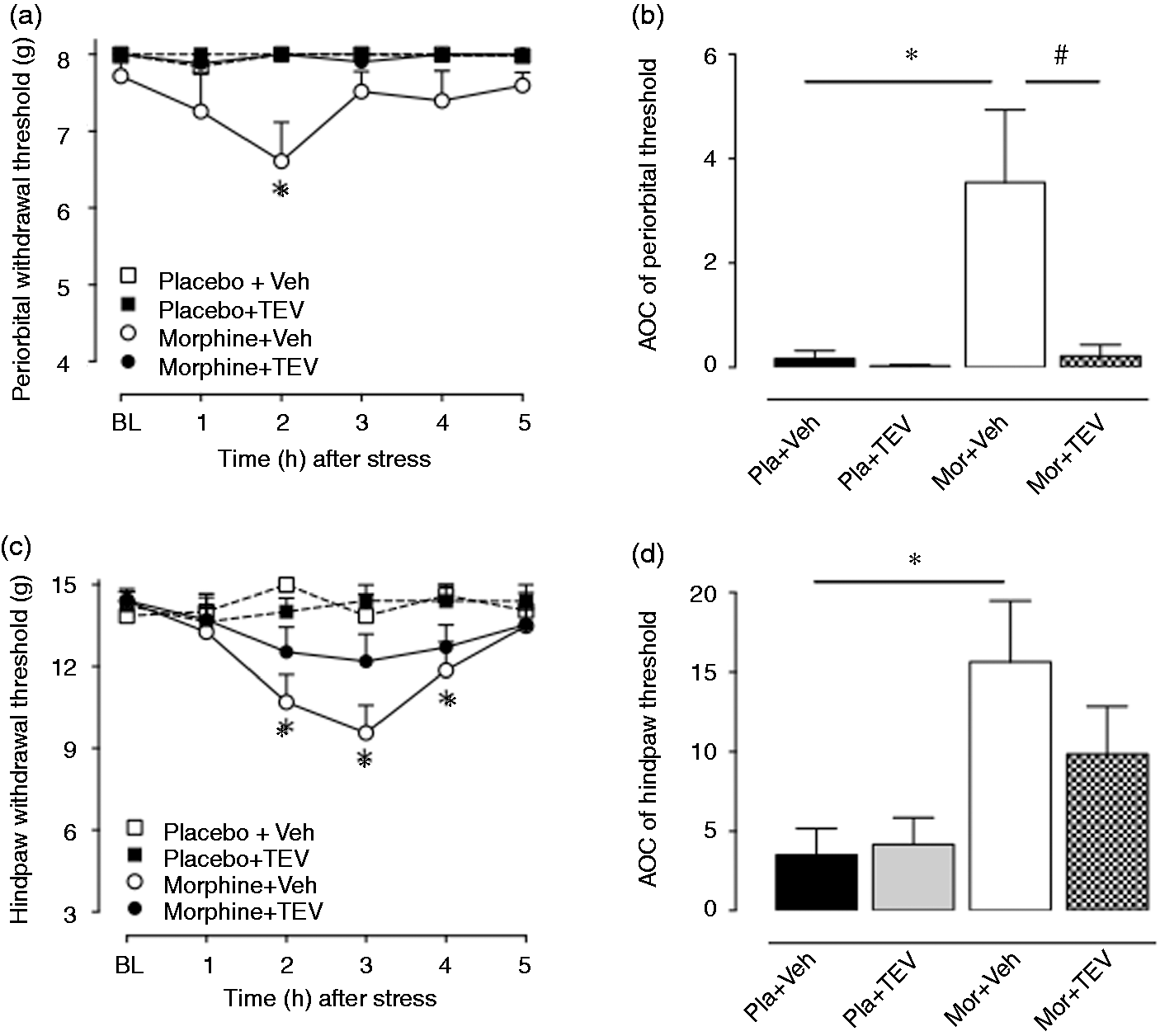
BLS increases plasma but not CSF CGRP levels in rats with sumatriptan-induced latent sensitization
Plasma and CSF CGRP levels were measured 30 minutes post-second BLS in sumatriptan and saline pre-exposed rats (Figure 5). A 62% increase in plasma CGRP was detected in sumatriptan pre-exposed rats compared to the saline-infused group (Figure 5(a) and 5(b); p < 0.05). A slight (24%) but not significant increase in CGRP was found in the CSF of sumatriptan pre-exposed rats compared to the saline group (Figure 5(c) and 5(d); p > 0.05).
Plasma and cerebrospinal fluid (CSF) calcitonin gene-related peptide (CGRP) levels in rats with sustained sumatriptan-induced latent sensitization post-bright light stress (BLS). On day 21 post-sumatriptan (0.6 mg/kg/day for 7 days subcutaneously) or its vehicle (saline, 1 ml/kg/day subcutaneously) infusion, jugular blood (plasma) and CSF were collected 30 minutes after the second BLS day exposure for 1 hour. (a) and (b) represent plasma CGRP concentrations and plasma CGRP concentration percentage controls, respectively. (c) and (d) represent CSF CGRP concentrations and CSF CGRP concentration percentage controls, respectively. Values are means ± SEM of 8–17 rats per group. Unpaired t-tests (two tailed) were applied in order to analyze the data.
Discussion
Although the events that initiate a migraine attack remain unknown, the activation of the trigeminovascular system is thought ultimately to be essential for migraine headache. Multiple lines of evidence support the conclusion that CGRP plays an important, but not exclusive, role in the generation of a migraine headache (5). Blood levels of CGRP, but not of substance P, are elevated during migraine (9), and CGRP provokes migraine in migraineurs (10), but not in normal volunteers (14). Inhibition of neurotransmitter release by the activation of 5HT1B/D presynaptic receptors on the peripheral and/or central terminals of trigeminal afferents is likely to contribute to triptan efficacy (15). CGRP receptor antagonists and anti-CGRP antibodies, respectively, are clinically efficacious for acute and preventative migraine treatment (6,16–22).
TEV48125 is a fully humanized anti-CGRP monoclonal antibody that is intended for the prevention of chronic and episodic migraine (18,19). Two recent Phase IIb trials demonstrated positive results in terms of efficacy, safety and tolerability for the preventative treatment of both episodic and chronic migraine with TEV48125 (11,18,23). In the present study, the potential prophylactic effect of TEV48125 was assessed in a rat model of MOH induced by pre-exposure to sumatriptan or morphine. We show that a single subcutaneous administration of the antibody prevented the generalized CA evoked by presumed migraine triggers in rats with medication-induced latent sensitization.
NO donors such as nitroglycerin and nitroprusside cause delayed headaches in migraineurs that are indistinguishable from a migraine attack (3,8,12). Precipitating migraine in susceptible individuals via the administration of nitroglycerin also causes increases in CGRP in the jugular venous blood (24). We have previously shown that challenge of sumatriptan-sensitized rats with a NO donor evoked delayed, generalized periorbital and hind-paw CA that was accompanied by increased plasma levels of CGRP (3). Administration of sumatriptan or morphine to uninjured rats increased the number of CGRP-positive, retrograde-labeled trigeminal ganglion neurons that projected to the dura mater, which was maintained for at least 3 weeks following discontinuation of the treatment (i.e., at day 21 after mini-pump or pellet implantation) (4). The increase in the number of CGRP-positive dural afferent cell bodies following triptan or opioid treatment may be related to the observed increase in CGRP blood levels following provocative stimuli such as SNP, although the source of CGRP in the blood has not been definitively established. These observations, however, do not allow for a conclusion to be drawn of a causal role of CGRP in the observed CA in this model. The finding in the present study that pretreatment 3 days before administration of the NO donor challenge with an anti-CGRP antibody prevented NO donor-induced CA provides strong support for this hypothesis.
While CGRP may contribute to migraine pain through peripheral mechanisms, this peptide also has prominent activity within central structures, including areas such as the TNC, thalamus, hypothalamus, ventral tegmental area and periaqueductal grey (25). The actions of CGRP within the central nervous system have been suggested to possibly contribute to migraine premonitory symptoms, as well as to other symptoms that are observed during migraine (26). However, whether the central actions of CGRP contribute to migraine pain is not clear. Previous reports in preclinical models induced by nitroglycerin demonstrated increased CGRP levels in the dura mater, as well as within the TNC, which were accompanied by increased expression of FOS-positive cells, suggesting neuronal activation within this structure (27). The nitroglycerin-induced upregulation of TNC FOS-positive cells was inhibited by pretreatment with olcegepant, a small-molecule CGRP receptor antagonist (26). These observations were supported by the work of Feistel and colleagues, who showed that the blockade of CGRP receptors by the non-peptide CGRP receptor inhibitor MK-8825 prevented the increase in activity of TNC neurons after continuous nitroglycerin infusion, suggesting that CGRP may be important in an early phase of nitroglycerin-induced central trigeminal activity (28). Thus, blockade of CGRP receptor signaling at both peripheral and central sites could contribute to the acute efficacy of small-molecule CGRP receptor antagonists. Further insight into this question has been provided by the discovery and development of [11C]MK-4232, a CGRP receptor positron emission tomography tracer that was used in humans to demonstrate that telcagepant achieved only low central receptor occupancy at efficacious doses (29). This result suggests that the peripheral actions of CGRP are sufficient to promote migraine pain, although whether increased brain penetration of CGRP receptor antagonists during migraine attacks contributes to their efficacy remains to be evaluated (29).
Monoclonal antibodies are generally considered not to cross the blood–brain barrier, and for this reason, TEV48125 is likely to act by targeting CGRP signaling in the periphery (30). In the present study, pretreatment with TEV48125 at 3 days prior to a provocative stimulus in rats primed by prior exposure to sumatriptan or morphine resulted in a significant inhibition of CA as induced by a NO donor or BLS. These observations suggest that peripheral CGRP neutralization by TEV48125 is sufficient to prevent the central amplification mechanisms that are necessary for the expression of delayed and generalized allodynia as initiated by migraine triggers and are consistent with the preliminary evidence of the clinical efficacy of TEV48125 for migraine prevention.
Our studies also showed that a single injection of TEV48125 at 30 mg/kg on day 18 following sumatriptan- or morphine-induced sensitization also blocked the development of CA induced by BLS, as assessed on day 21 following mini-pump implantation. Stress may be the most common migraine trigger and has been reported to be the primary trigger for migraine in 59% to over 80% of patients (31). Exposure to bright light was used as a stress paradigm in this animal model based on the well-known propensity of nocturnal rodents to prefer dark. It is unlikely that the bright light paradigm reflects photophobia, a symptom that is found in the majority of migraineurs (32). In our studies, we used BLS as an environmental stress that may initiate central and peripheral pathways, including sympathetic responses, which could lead to CA. Importantly, CA develops slowly after the termination of BLS, with a peak observed at approximately 2–3 hours, suggesting it acts predominantly as a stress trigger.
Although both the peripheral and central nervous systems produce CGRP, the main source of the stimulated release of CGRP is thought to arise from the primary afferents into the jugular blood and CSF (33). Consistent with this possibility, we found a significant increase of CGRP in plasma from the jugular blood after BLS in rats with sumatriptan-induced latent sensitization. BLS also produced a slight but not significant increase in CGRP levels in the CSF from the sumatriptan-sensitized rats. These findings are consistent with the observed blockade of BLS-induced CA via peripherally administered CGRP antibody, which is unlikely to penetrate the blood–brain barrier in the animal model. Additionally, these findings are consistent with clinical observations of increased CGRP in the jugular blood and the CSF of migraineurs (34), although it should be noted that we were unable to detect a significant difference in our treatment groups in terms of CSF levels. Collectively, the data suggest that the prevention of stress-induced CA likely reflects an inhibition of either CGRP-dependent activation and/or a sensitization of nociceptive afferents that generate and/or amplify migraine pain.
TEV48125 was given only 3 days prior to challenge with presumed migraine triggers. Longer pretreatment times and detailed time course studies were not performed due to the likely expression of antibodies against this humanized drug in rats. TEV48125 was well tolerated in cynomolgus monkeys, even when administered in high single or repeated doses for up to 14 weeks (35). That study also showed that neither the single nor repeated administration of high doses of TEV48125 resulted in blood pressure changes or electrocardiogram abnormalities (35), suggesting that these factors are unlikely to be significant in our studies in rats. Future studies with rodent-specific anti-CGRP antibodies may allow for more precise determination of the consequences of the long-term blockade of peripheral CGRP signaling on central changes induced by migraine-related events such as stress.
Conclusion
Peripheral CGRP neutralization can provide effective prevention of migraine-like pain in a rat model of MOH induced by prior exposure to sumatriptan or morphine. The data support a causal role for peripherally acting CGRP that follows common triggers of migraine, ultimately resulting in central adaptations that reflect a sensitized state.
Article highlights
Anti-CGRP monoclonal antibodies to the peptide, or to the receptor, may represent an effective and safe approach for the prevention and/or treatment of medication overuse headache (MOH). These data support the utility of this model of MOH for translational mechanistic studies and for screening existing and new treatments for a potential role in migraine prevention and the treatment of MOH.
Footnotes
Acknowledgements
The group sincerely thanks the CAPES agency for providing scholarship support for CMK. SW, JS, MB, DD and FP conceived and planned the study; CMK, JYX, NME and BR performed the experiments and analyzed the data; JYX, CMK and FP wrote the manuscript; JS, MB, JGC and DD corrected and improved the manuscript.
Declaration of conflicting interests
The authors declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: JS and MB are employees of Teva; SW was a former employee of Teva.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This study was funded by Teva through a grant to FP.