
Abstract
Introduction
Tolosa–Hunt syndrome (THS) is one of the most common ‘benign’ causes of painful ophthalmoplegia. Diagnosis is based on clinical and imaging findings and the exclusion of other causes because there is no specific biomarker for the syndrome. Eales disease, an idiopathic inflammatory venous disease that primarily affects the eye, can also affect the central (as stroke or myelitis) and peripheral nervous system.
Case report
We report the case of a 32-year-old woman with a subacute left ophthalmoplegia and evidence of a gadolinium-enhanced lesion suggesting an inflammatory granuloma that resolved within 48 hours after treatment with steroids. A diagnosis of THS was considered at this time. On a follow-up ophthalmological examination, a diagnosis of Eales disease with involvement of the left eye was made. The patient was treated successfully.
Conclusion
Eales disease could be a cause of painful ophthalmoplegia and may mimic THS. Long-term follow-up of patients diagnosed with THS may be necessary to exclude other diagnoses.
Introduction
Painful ophthalmoplegia (PO) consists of periorbital or hemicranial pain with paralysis of the ipsilateral ocular motor nerves. There is a variety of causes of this syndrome and neuroimaging using magnetic resonance imaging (MRI) is important in the detection of structural anomalies (1). Tolosa–Hunt syndrome (THS) and diabetic microvascular neuropathy are among the most frequent ‘benign’ causes of PO (2).
Eales disease (ED) is an idiopathic, usually painless, inflammatory venous occlusion that primarily affects the peripheral retina of adults (3). Perivascular phlebitis, peripheral non-perfusion and neovascularization are common retinal changes; bilateral involvement develops in 50–90% of patients. The disease predominantly affects young adult men (aged 20–30 years) and frequently manifests with decreased visual acuity, a consequence of recurrent vitreous haemorrhage (3). Although its aetiology seems to be multifactorial, mycobacterium species could have a role and a positive polymerase chain reaction for Mycobacterium is found in around 50% of patients with ED, but in only 10% of controls with other retinal vasculitis (3,4). Ophthalmological symptoms are the most frequent presentation of ED, but extra-ocular, in particular, neurological symptoms may be seen (5). To the best of the authors’ knowledge, PO has never been described as a manifestation of ED. We discuss a case of a patient with ED who presented with PO that was initially diagnosed as THS.
Case report
A 32-year-old white woman was admitted to our inpatient unit with left periorbital pain of three months’ duration, which improved on treatment with analgesia (paracetamol + codeine). Sometimes the pain was accompanied by an ill-defined sensation of blurred vision. About two and a half months after the onset of the pain, the patient developed diplopia when looking to the left. When evaluated, a left abducens paralysis and pin-prick hyposthesia in the left V1 territory was observed. The corneal reflex was bilaterally present. The patient reported a self-limited episode of diplopia with a duration of less than 10 days one year previously. No other relevant data of the previous episode was available. The patient reported no other previous relevant medical or surgical history.
MRI showed a 3 mm structure with gadolinium enhancement adjacent to the left abducens nerve, suggesting an inflammatory granulomatous lesion (Figure 1). No parenchymatous lesion was present. Investigations for an autoimmune (Antinuclear antibodies (ANA), Anti-neutrophil cytoplasmic antibody (ANCA), Extractable nuclear antigens (ENA), Angiotensin-converting Enzyme (ACE)) or infectious cause (Brucella sp., Syphilis sp., HIV, hepatitis C virus and hepatitis B virus) were normal or negative and normal protein (38 mg/dL), glucose (60%) and cell (2 cells/uL) levels were found in her cerebrospinal fluid, with negative Borrelia and neurotropic virus tests. No agent was identified on bacteriological study of her cerebrospinal fluid.
Brain magnetic resonance imaging scan performed before treatment with steroids (T1-weighted image with gadolinium contrast). Arrow: gadolinium enhancement of a lesion adjacent to the abducens nerve.
The patient was examined by a neuro-ophthalmologist and no pathological finding was present on fundoscopy. The patient was treated with intravenous methylprednisolone (1 g/day) for 5 days, with the resolution of the headache and diplopia in less than 48 hours. Although not fulfilling the time criteria for THS, in the absence of another diagnosis we managed the patient according to this working diagnosis and maintained her on a slow reduction regimen of the steroid treatment. The patient stopped steroid treatment after three months. No recurring or new symptoms were noted.
At the nine-month follow-up ophthalmological examination (performed by the same ophthalmologist, six months after stopping steroid treatment), signs of retinal periphlebitis with peripheral venous leakage and non-perfusion were noted in the left eye. A positive Mantoux test (enduration over 30 mm with central necrosis) and an interferon-gamma release array reaction were found. At this time a diagnosis of ED was established and the patient was managed accordingly for nine months with anti-tuberculosis drugs (isoniazid, rifampicin, pyrazinamide and ethambutol with pyridoxine supplementation during the first two months, with maintenance on isoniazid, rifampicin and pyridoxine for a further seven months) and retinal laser photocoagulation (Figure 2) with good tolerability. MRI was performed at this time and there was a regression of the previously noted lesion. No evidence of extra-ocular tuberculosis was found. No long-term neurological or ophthalmological impairment was evident at the two-year follow-up.
(a) Fluorescein angiography performed at follow-up showing peripheral venous leakage. (b) Pan-retinal photocoagulation.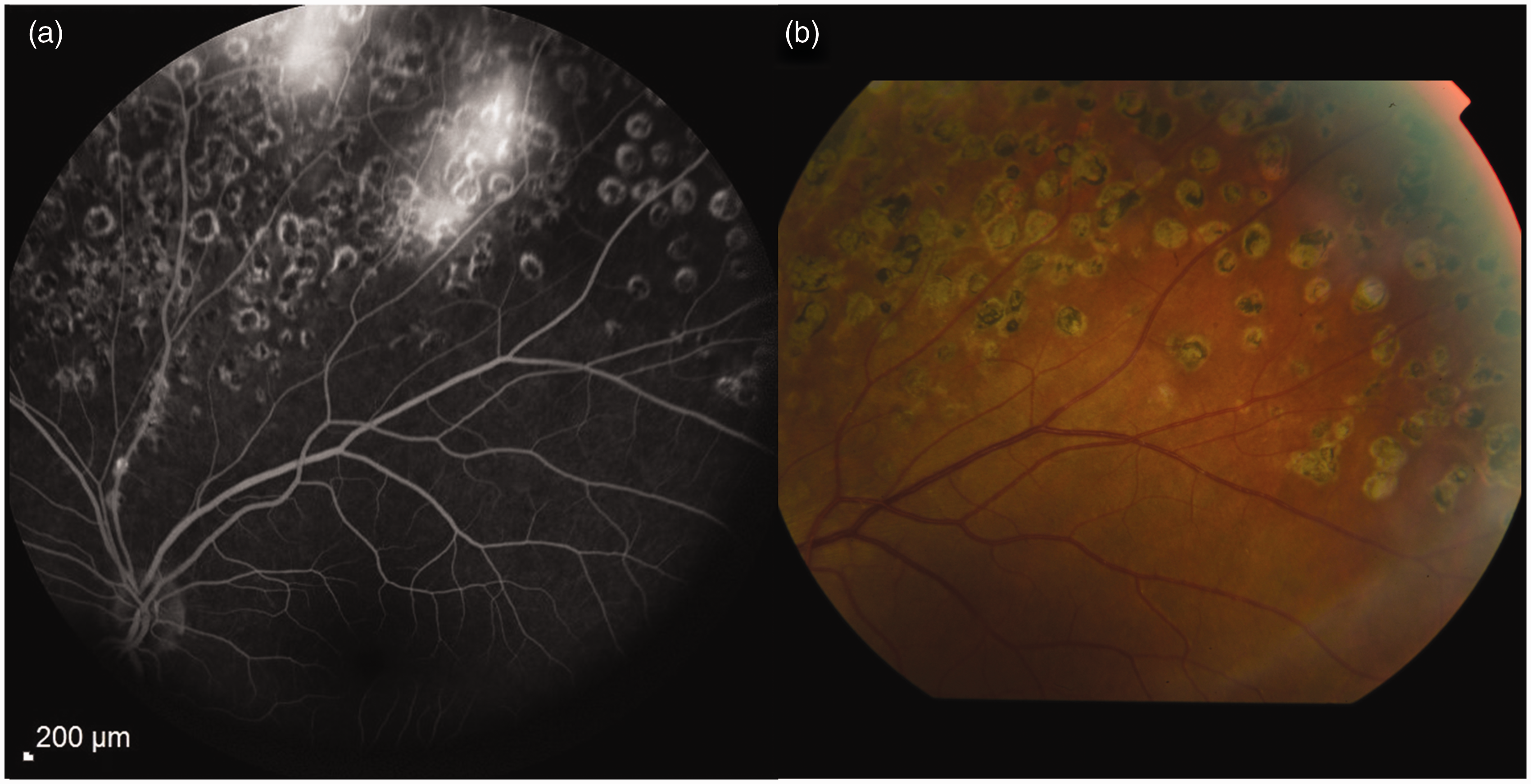
Discussion
Eales disease is a vasculopathy frequently diagnosed by retinal involvement. It is currently rare in developed countries, but is commonly reported in the Indian subcontinent with an incidence of 1 in 200–250 ophthalmic patients (3). There are no consensus criteria of ED; the disease is usually diagnosed by: (a) a characteristic clinical picture characterized by the presence of floaters, blurring of vision or a diminution in vision due to vitreous haemorrhage; and (b) findings from fluorescein angiography that delineate areas of non-perfusion, retinal and/or optic disc neovascularization, macular oedema or other changes resulting from retinal perivasculitis. The clinical manifestations are due to three basic pathological changes: inflammation (peripheral retinal perivasculitis); ischaemic changes (peripheral retinal capillary non-perfusion); and neovascularization of the retina or disc, which often leads to vitreous haemorrhage (6).
Neurological causes are the most common extra-ocular manifestations. These include myelitis or myelopathy (5,7–9), stroke (5,10–12), internuclear ophthalmoplegia (13), ataxia (9), peripheral neuropathy (14) and cranial nerve paralysis (15). The neurological manifestations of ED are usually responsive to steroids and the ophthalmological symptoms precede neurological symptoms in more than 50% of patients by a period of six months to 10 years (5).
Steroid responsiveness is also a common feature of THS. This was a diagnostic criterion in previous versions of the ICHD, but was downgraded in the recent ICHD-III beta criteria. Even with the downgrade, the usefulness of this response for diagnosis is well documented on previous research and in the comment section of ICHD-III beta (16).
THS is a frequent cause of PO and, in the absence of clear biomarkers, it is often considered to be a diagnosis of exclusion (i.e. not better accounted for by another diagnosis) (16). The diagnostic criteria of THS, according to ICHD-III beta, imply the presence of a unilateral headache around the brow and eye preceding ipsilateral ophthalmoparesis by less than two weeks. The ophthalmoparesis should involve one or more of the ipsilateral third, fourth and/or sixth cranial nerves; the presence of a granulomatous inflammation of the cavernous sinus, orbit or superior orbital fissure should be demonstrated by MRI or a biopsy sample (16).
With the presence of a steroid-responsive PO with evidence of granulomatous inflammation of the orbit, and in the absence of a working alternative, our patient was initially diagnosed with THS. Although there was a larger temporal interval between the start of the headache and the onset of ophthalmoplegia than defined by the standard criteria, these time criteria were defined on expert consensus and previous studies have found that more than 10% of patients with probable THS had time intervals longer than the defined two weeks between symptoms (16,17).
In the only series that described post-mortem findings for nine patients with ED with central nervous system involvement, ED was described as an inflammatory venopathy with perivenular demyelination (14,18). Similarly, in a report of a 38-year-old woman with ED who had bilateral stroke, the angiography revealed beading of the Sylvian arteries (11). Vascular mechanisms are probably related to the neurological manifestations of this disorder. Although the pathological mechanisms of ED are currently not clearly established, we hypothesize that a chronic inflammatory microangiopathy of the periorbital vasculature was the cause of the pain in our patient and when it involved the left abducens and ophthalmic nerves vasa nervorum, it caused the ophthalmoplegia. The steroid treatment probably reversed the inflammatory vascular changes, leading to an improvement in the clinical symptoms.
We believe it is important to carry out long-term follow up of patients with probable THS as different diagnosis could emerge, as with our patient. This follow-up should include a complete ophthalmological examination. Also longer time periods between the onset of headache and ophthalmoplegia, even not excluding THS, should also raise a suspicion for a different cause of the PO; this should be kept in mind after the acute phase has passed.
Based on one simple case report, no clear link can be established of a causal relationship between steroid-responsive PO and ED in this patient. Nevertheless, we found it reasonable to consider the presence of a link between them as PO and retinopathy were on the same side with a clear temporal relationship, cranial neuropathies have previously been described in ED, and neurological manifestations of ED usually respond to treatment with steroids. We found it less probable that the patient had an atypical presentation of THS with ED diagnosed at early follow-up. To our knowledge, this is the first report of ED as a cause of steroid-responsive PO.
The ICHD-3 criteria are currently published in a beta version to allow field testing and revision before the final version is published. We hope that some of these considerations are taken into account in the final definition of the criteria for THS (17).
Clinical implications
Eales' disease could be a cause of steroid-responsive painful ophthalmoplegia mimicking Tolosa–Hunt syndrome. In patients with suspected Tolosa–Hunt syndrome, a detailed ophthalmological examination should be performed and, in atypical Tolosa–Hunt syndrome, this should be complemented with fluorescein angiography. Long follow-up of patients with Tolosa–Hunt syndrome may be necessary to exclude other diagnoses.
Footnotes
Declaration of conflicting interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.