
Abstract
Tolosa-Hunt syndrome (THS) is characterized by unilateral painful ophthalmoplegia with oculomotor paresis, associated with an idiopathic granulomatous inflammation involving the cavernous sinus, with a typical relapsing-remitting course. We report a case of an 8-year-old girl who was admitted because of an ophthalmoplegia with exotropia and ptosis of the left eyelid, accompanied by diplopia and left sovraorbital pain. The clinical data, neuroradiological findings and response to steroid treatment suggested THS, as defined by the 2004 International Classification of Headache Disorders (ICHD)-II criteria. THS must be considered a possible cause of painful ophthalmoplegia in childhood, as well as in adults, and confirmed with a focused neuroradiological investigation. The few paediatric cases described in the literature that meet the 2004 ICHD-II criteria are not sufficient to identify possible differences between the paediatric and the adult forms. Every new paediatric case should therefore be reported in order to gather and compare further information.
Introduction
Tolosa–Hunt syndrome (THS) is the label coined by Smith and Taxdal in 1966 to identify an ophthalmoplegia characterized by unilateral periorbital pain and oculomotor paresis. The syndrome was first described in 1954 by Tolosa (1) in a patient with unilateral orbital pain and ophthalmoplegia. The patient's cerebral angiography showed suggestive narrowing of the carotid siphon, slightly distal to the cavernous sinus, while post-mortem studies indicated a granulomatous inflammation of the affected cavernous sinus and carotid artery. Some years later, in 1961, Hunt et al. (2) described six similar patients and underlined the prompt steroid responsiveness among the principal features of the syndrome.
The International Headache Society, for the first time in 1988 (3) and subsequently in 2004 (4), included THS among the ‘neuralgias’, defining the following five diagnostic criteria:
One or more episodes of unilateral orbital pain, persisting for weeks if untreated
Paresis of one or more of the third, fourth and/or sixth cranial nerves and/or demonstration of granuloma by magnetic resonance imaging (MRI) or biopsy
Paresis coincides with the onset of pain or follows it within 2 weeks
Pain and paresis resolve within 72 h when treated adequately with corticosteroids
Other causes excluded by appropriate investigations.
The first and most important clinical monograph on THS is that of Bruyn and Hoes (5), who gathered and reviewed the data of > 300 cases from 1954 to 1986.
Later, La Mantia et al. (6) examined and reviewed all cases published between 1988 and 2002, on the basis of the new International Classification of Headache Disorders (ICHD)-II 2004 criteria. The authors concluded that the application of the clinical criteria alone does not guarantee correct diagnosis and that MRI is mandatory. They underlined the need for MRI criteria (in terms of acquisition protocol and follow-up timing) to demonstrate the inflammatory lesion and its resolution after treatment.
The most recent study is that of Colnaghi et al. (7), who conducted a bibliographical review of papers published on THS between 1999 and 2007. The authors confirmed the central role of MRI in the diagnosis of this pathology and emphasized the need for more data about the steroid dosing schedule and the timing of clinical resolution, differentiating pain and paresis.
As in adults, THS also represents a possible cause of painful ophthalmoplegia in childhood. Few cases of THS have been described in the paediatric population, particularly after the introduction of the ICHD-II criteria in 2004. In this paper we report a new case that fulfilled these diagnostic criteria.
Case report
V.G., an 8-year-old girl, was admitted to hospital for acute onset of dull supraorbital pain, diplopia, exotropia and ptosis of the left eyelid. A week prior to admission, she suffered from abdominal pain, diarrhoea and vomit.
On admission, physical examination revealed left ophthalmoparesis due to deficits of the third and fourth cranial nerves, with ptosis of the left eyelid. Other neurological and physical examinations were normal. The child's personal and family history of headache or migraine was negative. The fundoscopic examination was also normal.
Complete blood count, erythrocyte sedimentation rate and C-reactive protein screen were average.
Several laboratory investigations were performed in order to exclude the main causes of symptomatic ophthalmoplegia, such as systemic autoimmune disease or infective processes. These examinations yielded negative results and included: hepatic enzymes, coagulation tests, creatinine, urea, ammonium, C3 and C4 complement factors, antinuclear antibodies [anti-dsDNA, perinuclear antineutrophil cytoplasmic antibody (ANCA), cytoplasmic ANCA], lupus erythematosus cell preparation, anti-thyroglobulin antibodies, antithyroid peroxidase antibodies, antimitochondrial antibodies, and anti-Borrelia antibodies.
Lumbar puncture revealed average opening pressure, with normal cell count, protein and glucose, and sterile fungal and bacterial cultures. The EEG was also normal.
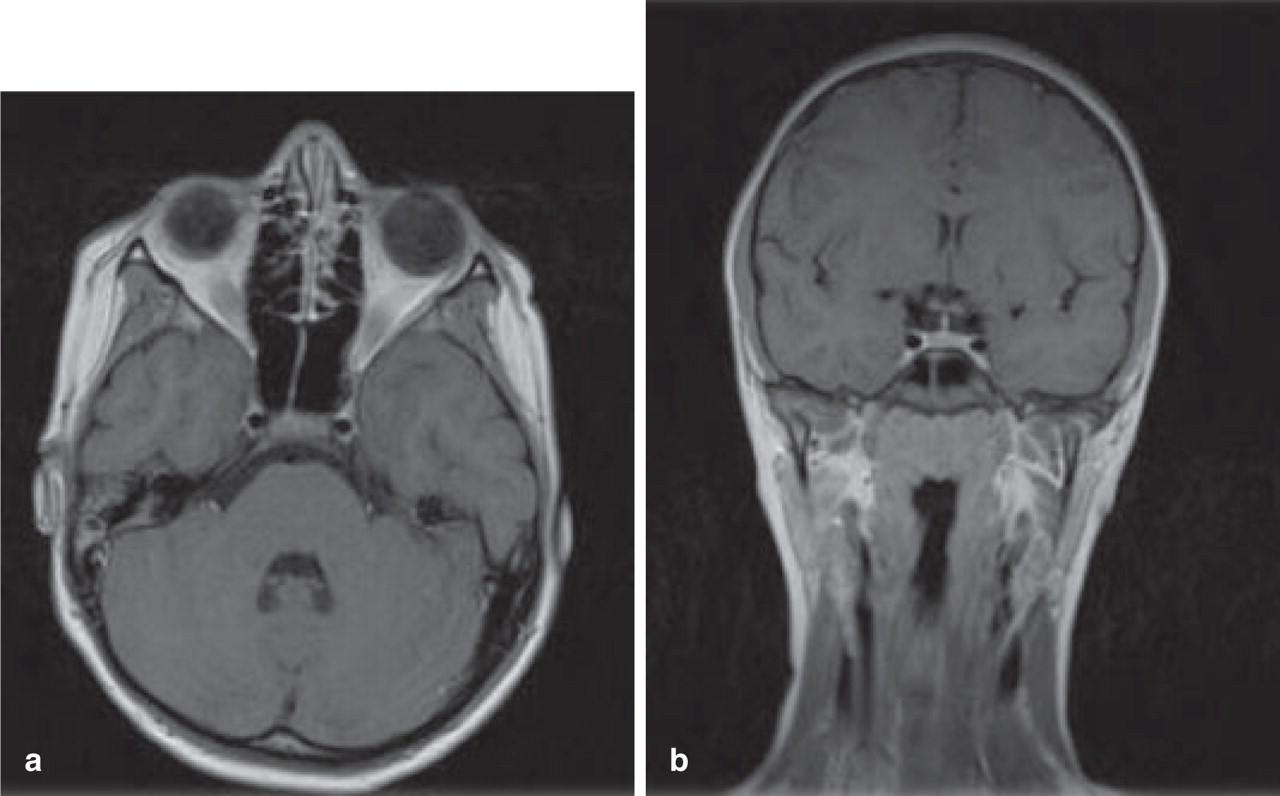
MRI showed mild asymmetry of the cavernous sinus and the presence of a hypointense tissue, homogeneously enhancing at contrast administration; the lesion was approximately 3 mm thick and situated laterally and caudally to the anterior sectors of the intracavernous portion of the carotid artery (Fig. 1). There were no abnormalities of the other structures of the orbit, and a sphenoid sinusitis was excluded.

(a) Spin-echo (SE) T1-weighted post-gadolinium axial magnetic resonance (MR) image: enlargement of left cavernous sinus secondary to a soft tissue mass that enhances homogeneously after contrast injection. The calibration of the intracavernous portion of the left internal carotid artery is decreased compared with the other side. (b) SE T1-weighted post-gadolinium coronal MR image.
The association of clinical data and MRI findings, as well as the exclusion of other possible causes, led to the diagnosis of THS.
Corticosteroid treatment was immediately started (dexamethasone 0.2 mg/kg per day) and continued for 8 days. Significant pain relief was observed within 72 h, while the ophthalmoparesis required about 2 weeks for resolution.
Six months later, the MRI showed complete resolution of the neuroradiological signs (Fig. 2).
(a) Spin-echo (SE) T1-weighted post-gadolinium axial magnetic resonance (MR) image: after 6 months MR images show complete regression of the soft tissue mass and normal calibration of the left internal carotid artery. (b) SE T1-weighted post-gadolinium coronal MR image.
At the 2-year follow-up, no clinical recurrence was reported and the MRI normalization was confirmed (Fig. 3).
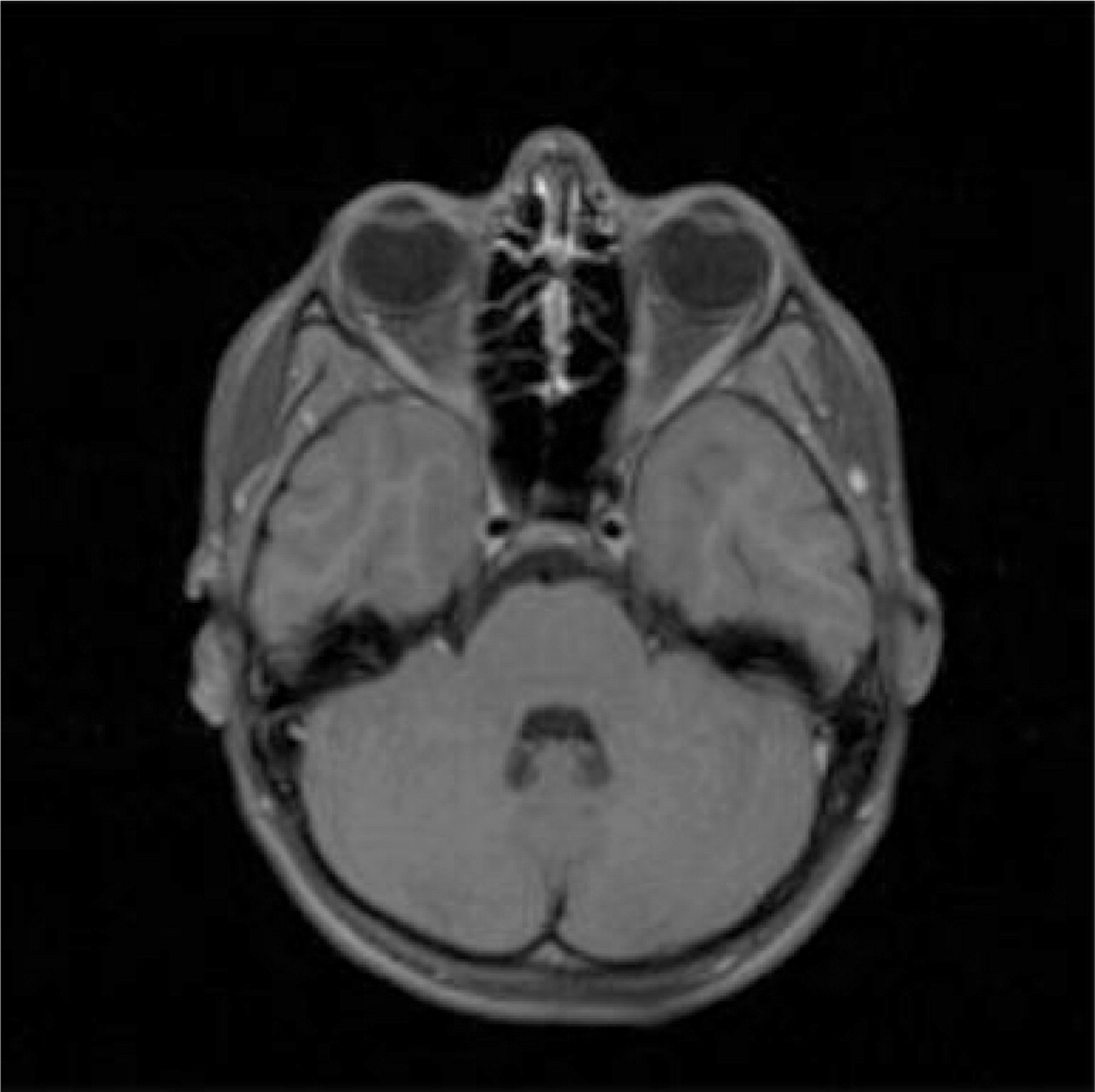
Spin-echo T1-weighted fat-suppressed post-gadolinium axial magnetic resonance image: after 2 years complete regression of the soft tissue mass and normal calibration of left internal carotid artery persist.
Discussion
There is general agreement that the clinical features suggesting THS are not specific to this syndrome but are common to several conditions, so their presence alone is not sufficient for a correct diagnosis and a neuroimaging study is mandatory (6, 7). At present, the most sensitive technique in revealing the signs of the typical granulomatous lesion involving the cavernous sinus is MRI, performed so as to provide specific views of the cavernous sinus and orbits, including pre- and post-gadolinium images. The importance of MRI in the diagnosis of THS was first underlined and confirmed by La Mantia et al. (6). In a critical review of the literature on THS published from 1988 to 2002, the authors concluded that only 44 of the 208 cases reported in that period met the required MRI criteria and could have actually been classified as THS according to the ICHD-II 2004 criteria (providing imaging or bioptic evidence of granuloma). The authors hypothesized that more cases would have been diagnosed with a better defined neuroimaging protocol, whereas the diagnosis of THS with negative MRI findings remains an unresolved issue. More recently, Colnaghi et al. (7) specified which MRI techniques should be used to demonstrate the presence of inflammatory tissue, thus reducing the possibility of false negatives: 3 mm thickness, coronal and axial T1 spin-echo and T2 turbo spin-echo sequences, T1 fat-suppressed at the orbit and cavernous sinus level. Moreover, they emphasized the importance of the localization and extension of the inflammatory tissue on MRI as a prognostic factor: they distinguished different diagnostic groups and observed that when the inflammation extends beyond the cavernous sinus and the orbit, pain responsiveness to steroid treatment is similar but with probably a higher frequency of recurrence.
Referring to criterion D of the ICHD-II for THS (‘pain and paresis resolve within 72 h when treated adequately with corticosteroids’), Colnaghi et al. (7) observed that there is no evidence as to what kind of steroid treatment is adequate and emphasized that the 72-h time limit should be applied only for pain resolution. Consequently, they propose rewriting the diagnostic criterion D as ‘pain resolves within 72 h when treated with corticosteroids’.
In paediatric literature, only few cases of THS have been reported. Since the Bruyn literature survey (5), < 40 cases have been described, most of which cannot be confirmed because they do not meet the ICHD-II 2004 standards, since they lack the MRI criteria. This is true of all those cases reported prior to the introduction of MRI into clinical practice, such as the two cases reported by Terrence and Samaha in 1973 (8) and considered the first in paediatric literature. The paediatric literature until 2004 also includes several cases clinically diagnosed as THS that had symptoms of other conditions, such as meningioma (9), sphenoid sinusitis (10), lymphoma (11) or tuberculous pachymeningitis (12).
In 2001, Del Toro et al. (13) reported the case of a 10-year-old boy who presented a first episode of retro-orbital pain that lasted for a few days, followed, about a month later, by diplopia and ocular pain. The authors described a complete right third-nerve palsy with lateral deviation of the right eye, inability to adduct, lift and lower the eye, a dilated pupil sluggishly reactive to light, and ptosis of the eyelid. MRI revealed a right cavernous sinus enlargement, more visible after intravenous gadolinium administration, with stenotic internal carotid artery, confirmed by magnetic resonance angiography (MRA). Clinical remission with complete regression of neuroradiological signs spontaneously occurred in approximately a month. In this case ICHD-II criterion D (response to steroid treatment) was not respected.
The typical steroid responsiveness was described in the paediatric case reported by Koul and Jain in 2003 (14): after the administration of prednisolone 1 mg/kg per day the pain subsided within 72 h and the neurological signs regressed after 3 weeks; complete MRI normalization was observed after about 6 weeks.
In 2004, Kóbor et al. (15) reported the case of a 12-year-old girl with an intermittent clinical history of right periorbital pain and sixth nerve palsy, yet with initially ambiguous MRI and MRA findings; oral methylprednisolone was prescribed, but was discontinued after about a week because of its inefficacy. Five months later, a new MRI investigation revealed a 7-mm contrast enhancing hyperintense lesion (coronal T1-weighted MRI) along the intracranial segment of the internal carotid artery, bulging medially in the right cavernous sinus. The authors confirm the central role of neuroimaging, but also point out that the inflammatory signs may initially be undetectable by MRI. Therefore, they strongly underline the importance of clinical criteria, especially in cases with negative MRI, and the appropriateness of repeated MRI investigations, as well as close clinical and radiological follow-up. Moreover, the issue regarding steroid responsiveness needs to be clarified in terms of dosage and timing, as previously underlined (7).
Yeung et al. (16) described the case of a 9-year-old boy with acute-onset frontal headache, left orbital pain and complete third and fourth nerve palsy, with a mild left central scotoma. Steroid therapy with dexamethasone (1 mg/day) was taken for 2 weeks, with efficacy only on the pain. The MRI performed at that moment suggested THS (T1 isointense, T2 hyperintense lesion in the left cavernous sinus, intensely enhanced by contrast, with compression of left internal carotid artery and third cranial nerve). High-dose steroid therapy (prednisolone 2 mg/kg per day) was started, with complete resolution of the neurological signs within 2 weeks, and the MRI follow-up showed progressive reduction of the lesion. The authors observed that only high doses of steroids seemed to be effective. Nevertheless, they underlined the absence of defined dosage and timing of therapy in paediatric literature. Moreover, they emphasized the need for serial MRI to demonstrate complete normalization, define the response to therapy and to confirm the diagnosis.
Lastly, Orssaud et al. (17) reported and compared two cases of painful ophthalmoplegia. One of the two cases was diagnosed with THS because of the clinical features and the absence of a personal or family history of migraine; corticosteroid treatment (1 mg/kg per day) was prescribed, with rapid pain relief and progressive resolution of the oculomotor palsy. Although neuroimaging findings were negative, the authors concluded that the clinical data and the efficacy of steroid treatment were sufficient for diagnosis.
MRI negativity remains a topic of discussion, and many studies actually include a large number of negative MRI cases, like the 33% of La Mantia et al.'s review (6).
Conclusion
The case we report meets all the ICHD-II diagnostic criteria (4). Our young patient presented acute-onset unilateral ocular pain in association with paresis of the III and IV cranial nerves (criteria A and C). The specific MRI technique adopted revealed the typical radiological signs, with enlargement of the left cavernous sinus due to the presence of a soft tissue mass homogeneously enhancing after contrast injection (criterion B). A prompt steroid responsiveness was observed, with pain relief within 72 h and ophthalmoparesis resolution in about 2 weeks (criterion D). Other causes of painful ophthalmoplegia were excluded by appropriate investigations (criterion E). MRI follow-up performed after 6 months and after 2 years demonstrated complete resolution of the neuroimaging findings.
THS should be considered among the causes of painful ophthalmoplegia in childhood as well as in adults, and a focused neuroradiological investigation should be performed for correct diagnosis (6, 7, 12, 18).
In our case, the clinical course confirmed that the pain seems to resolve more rapidly than the ophthalmoplegia, also in children. The precociously administered medium/high dose of dexamethasone was effective, and at the 2-year follow-up no recurrence was reported. According to the diagnostic criteria (4) and clinical evidence, the response to steroid treatment represents an important component in the diagnostic process, but in paediatric literature, at present, there are no defined protocols regarding dosage and timing for comparison. In adults, the literature reports a risk of recurrence of 40%, with large variability over time and usually a milder clinical condition, owing to the fact that the diagnosis is reached and treatment is started earlier (19). Paediatric data are insufficient on this issue also.
In conclusion, each new paediatric case should be promptly reported in order to provide and compare more data. This could facilitate the identification of possible differences between the paediatric and adult forms and lead to a clearer definition of the specific characteristics of THS in childhood.