
Abstract
Background: Prostaglandin E2 (PGE2) has been suggested to play an important role in the pathogenesis of migraine. In the present experiment we investigated if an intravenous infusion of PGE2 would induce migraine-like attacks in patients with migraine.
Methods: Twelve patients with migraine without aura were randomly allocated to receive 0.4 µg/kg/min PGE2 (Prostin®E2, dinoprostone) or placebo over 25 minutes in a two-way, crossover study. Headache intensity was recorded on a verbal rating scale, middle cerebral artery blood flow velocity (VMCA) was measured by transcranial Doppler (TCD) and diameter of the superficial temporal artery (STA) was obtained by c-series scan (Dermascan C).
Results: In total, nine migraine patients (75%) experienced migraine-like attacks after PGE2 compared to none after placebo (p = 0.004). Seven out of 9 (58%) patients reported the migraine-like attacks during the immediate phase (0–90 min) (p = 0.016). Only two patients experienced the delayed migraine-like attacks several hours after the PGE2 infusion stop (p = 0.500). The VMCA decreased during the PGE2 infusion (p = 0.005) but there was no significant dilatation of the STA (p = 0.850).
Conclusion: The migraine-like attacks during, and immediately after, the PGE2 infusion contrast with those found in previous provocation studies, in which the other pharmacological compounds triggered the delayed migraine-like attacks several hours after the infusion. We suggest that PGE2 may be one of the important final products involved in the generation of migraine attacks.
Introduction
Prostaglandin E2 (PGE2) is a principal pro-inflammatory prostanoid involved in various biological functions, including pain processing (1–4). Experimental studies in animals (5) and in vitro studies on human tissues (6,7) suggest that PGE2 may play an important role in the pathogenesis of migraine. It has been reported that PGE2 is released from rat dura mater following the electrical and chemical stimulation in vitro (8). Furthermore, PGE2 stimulates release of calcitonin gene–related peptide (one of the key neuropeptides involved in migraine) in rat trigeminal neurones (9) and trigeminal nucleus caudalis (10) Studies in migraineurs have reported increased levels of PGE2 in saliva (11) and blood (12) during migraine attacks.
Over the past six years we have systematically evaluated the possible role of prostanoids, including PGE2, in headache and migraine in human provocation experiments (13–15). As a first step, we have studied whether PGE2 may provoke head pain in 11 healthy volunteers (14). Intravenous infusion of PGE2 induced head pain in all participants and, interestingly, one subject reported throbbing pain, pain aggravation by cough, nausea and photophobia (14). Whether PGE2 triggers migraine attacks in patients with migraine is unknown. We hypothesised that PGE2, similar to other pharmacological triggers (including prostaglandin I2) of migraine, would induce immediate non-migraine headache after infusion followed by delayed migraine-like attacks several hours after stop of infusion. To test this hypothesis we investigated migraine-provoking abilities of a naturally occurring PGE2 (Prostin®E2, dinoprostone) in a double-blind, placebo-controlled, crossover study in patients with migraine without aura.
Design, procedure and methods
Migraine patients (aged 18–55) were recruited via the web site www.forsøgsperson.dk and via the Danish Headache Center. Female participants were requested to have sufficient contraception. Exclusion criteria were any other type of headache (except episodic tension-type headache >3 per month), previous serious somatic, psychiatric or infectious diseases, or intake of daily medication including prophylactic migraine treatment. Physical, neurologic examination and electrocardiography (ECG) were performed on the day of enrolment.
The study protocol was approved by the Ethics Committee of the Country of Copenhagen (VEK H-B-2007-053) and the Danish Data Protection Agency, and performed in accordance with the Helsinki Declaration of 1964, as revised in 2008. The study was registered on www.clinicaltrials.gov. All patients gave their written informed consent.
In a double-blind, placebo-controlled, two-way, intra-individual crossover study, 12 migraine patients without aura were randomly assigned to receive 0.4 µg/kg/min of PGE2 (Prostin®E2, dinoprostone) (Pfizer Ltd., United Kingdom) or placebo (saline) intravenously over 25 minutes on two days separated by at least one week. Randomization and preparation of the study drug was done by medical stuff not involved in the study. The randomization code was kept in the hospital during the study and the unblinding procedure was first performed after the study was completed.
The patients were not allowed to smoke or consume any alcoholic beverages, caffeine, cocoa or chocolate for at least eight hours before the infusion. Use of pharmacologic agents apart from oral contraceptives was not permitted. The patients reported to the laboratory at 8:30 a.m. and were confirmed to have been headache-free for a minimum of 24 hours and migraine-free for at least 48 hours. Baseline values for flow velocity in the middle cerebral artery (VMCA), diameters of superficial temporal artery (STA) and radial artery (RA), mean arterial blood pressure (MAP), heart rate (HR), end-tidal partial pressure of pCO2 (PetCO2), transcutaneous arterial oxygen saturation (SAT), ECG, headache score and any adverse events (AEs) were recorded 30 minutes after application of an intravenous catheter (Venflon® [Becton, Dickinson, Helsingborg, Sweden]) and rest in the supine position. The procedures were performed in a quiet room at room temperature between 24.0°C and 24.7°C. The patient remained in the supine position throughout the study period from time −10 min (T−10) (start of study period) to T90 (end of study period, 65 min post-infusion). Following the baseline (T−10 and T0) measurements, subjects were randomized to PGE2 or placebo. At T0, the infusion was initiated by a time- and volume-controlled infusion pump (Braun Perfuser, Melsungen, Germany). All measurements were recorded at T−10 and then every 10 minutes until T90.
MAP and HRwere measured using an auto-inflatable cuff and PetCO2 was obtained with an open mask without any respiratory resistance (ProPac Encore®; Welch Allyn Protocol, Beaverton, OR, USA). ECG was recorded continually using Cardiofax 1550 (Nihon Cohden, Tokyo, Japan). VMCA was recorded by transcranial Doppler (TCD) ultrasonography (2 MHz) with handheld probes (Multidop X; DWL, Sipplingen, Germany); we used an average of four cycles, each lasting four seconds (16).
A high-resolution ultrasound scanner, c-series scan (20 MHz, bandwidth 15 MHz; Dermascan C; Cortex Technology, Hadsund, Denmark) was used to measure the diameter of the frontal branch of the left superficial temporal artery (STA) and the left radial artery (RA).
All measurements were done by the same skilled study personnel (LE and WG).
Headache intensity and adverse events
To record headache intensity we used a 10-point verbal rating scale (VRS), where 0 indicated no headache; 1, a very mild headache (including pre-pain—a pressing or throbbing feeling); 5 indicated moderate headache and 10 indicated the worst-imaginable headache (17). The patients were encouraged to self-report any changes in their well-being during the study and were questioned about the presence of headache, accompanying symptoms according to the International Classification of Headache Diseases, second edition (IHCD-II) (18) as well as adverse events (AEs) every 10 minutes from T−10 to T90. During the out-hospital phase, defined as a period after discharge and until bedtime, all patients were carefully instructed to make hourly recordings of headache and accompanying symptoms according to IHCD-II (18) and any other AEs. All AEs were classified as related or not related to the study drug by the investigator.
Migraine definition
Experimental migraine is not spontaneous and therefore cannot fulfil strict International Headache Society (IHS) criteria for migraine without aura. (18) The majority of patients report them as attacks that mimic spontaneous migraine attacks (15,19). However, it is well known that many spontaneous migraine attacks develop in a matter of hours and in the early stage phenomenologically only fulfil the criteria for tension-type headache and associated symptoms; unilateral localisation and increase in pain severity occur later. Furthermore, the majority of migraineurs can often predict development of migraine at the very early stage and cannot be denied treatment of experimentally induced headache. Based on these circumstances the following criteria were used to characterize migraine-like attack induced 0–12 hours after the infusion of Prostin® E2.
Migraine-like attack fulfilling IHS criteria 1 or 2:
Headache fulfilling criteria for migraine without aura C and D (18):
Headache has at least two of the following characteristics:
Unilateral location Pulsating quality Moderate or severe pain intensity (moderate to severe pain ≥4 on VRS) Aggravation by cough (in-hospital phase) or causing avoidance of routine physical activity (out-hospital phase) (e.g. walking or climbing stairs) During headache at least one of the following:
Nausea and/or vomiting Photophobia and phonophobia Headache described as mimicking usual migraine attack and treated with a triptan
Statistics
Headache intensity scores are presented as median and quartiles. Vascular variables are presented as mean ± standard deviation (SD). The maximum percentage change in diameter of the MCA from baseline is presented as mean ± SD.
Immediate headache or migraine-like attack was recorded during the in-hospital phase (0–90 min) and delayed headache or migraine-like attack was recorded during the out-hospital phase (1.5–12 h).
The primary end-points were differences in the incidence of migraine-like attacks and the area under the curve (AUC) for headache score (AUC0–90 min and AUC1.5–12 h) between PGE2 and placebo days. The secondary end-points were differences in the AUC for VMCA (AUCVMCA), STA (AUCSTA), RA (AUCRA), PetCO2 (AUCPetCO2), MAP (AUCMAP) and HR (AUCHR) between two experimental days. The data were baseline-corrected and the AUC for the time period T0–T90 for headache score, VMCA, MAP, HR and PetCO2 was calculated, using the trapezium rule (20). Assuming that no changes in cerebral blood flow (CBF) occur during the infusion (14) the maximum percentage change in diameter (Δd) of the MCA can therefore be calculated as: Δd = (√(VMCAtime0/VMCAtime20) −1 ) × 100 (21).
To test the statistical difference between the variables, we applied the McNemar test for nominal data (incidence of migraine like attacks, headache and AEs), the Wilcoxon signed ranks test for AUC for headache, and a paired, two-way t-test for vascular data.
Five percent (p < 0.05) was accepted as the level of significance. All analyses were performed with PASW Statistics 18 for Windows (SPSS Inc., Chicago, IL, USA).
Results
We enrolled 14 patients with migraine without aura (18). Two patients were excluded: one patient completed the first experimental day (PGE2) but did not report on the second day (we lost contact with this patient); one patient wished to abort the infusion after 10 minutes on day 2 (PGE2 day) due to severe nausea and discomfort. The excluded patients were not included in the statistical analysis. Thus, 12 patients with migraine (9 F/3 M, mean age 29.8 (range 19–52 years), mean weight 67.7 kg (range 57–82 kg) completed the study. Only one male patient has previously participated in a migraine provocation study conducted by our group.
Mean baseline values (±SD) of variables.
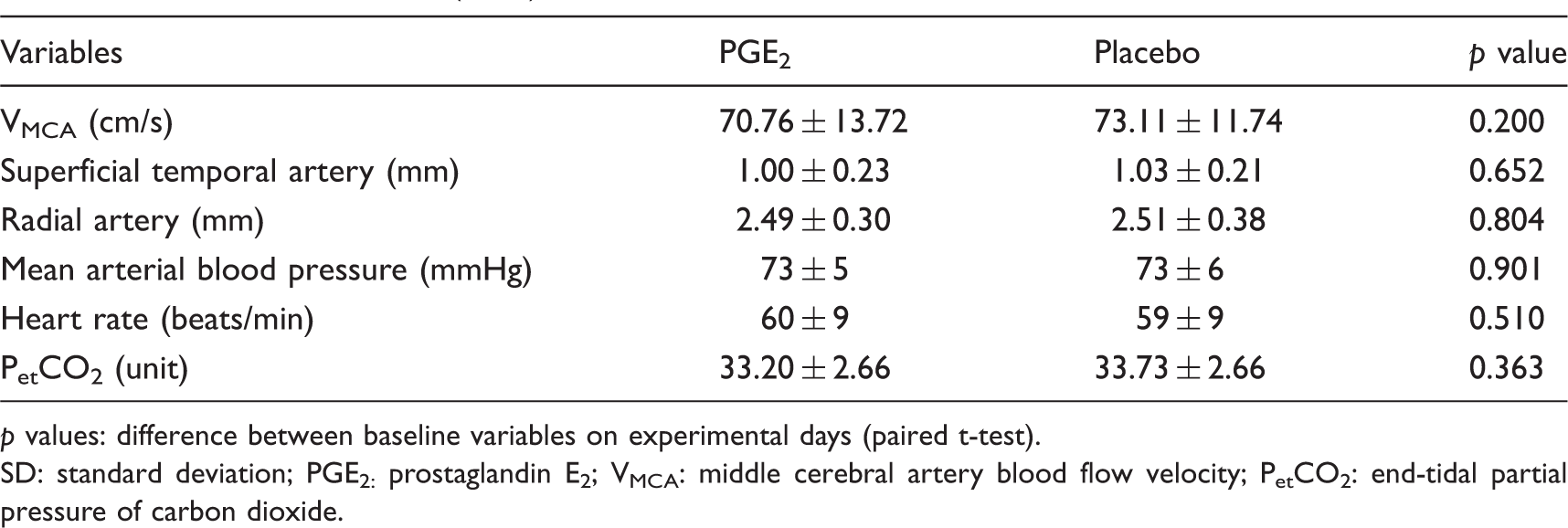
p values: difference between baseline variables on experimental days (paired t-test).
SD: standard deviation; PGE2: prostaglandin E2; VMCA: middle cerebral artery blood flow velocity; PetCO2: end-tidal partial pressure of carbon dioxide.
Adverse events recorded during the in-hospital phase after infusion of PGE2 and placebo.

There was an increased incidence of stiff muscles, increased saliva production, flushing, palpitations, heart sensation and paraesthesias on PGE2 day compared with placebo (p < 0.05, McNemar’s test).
There was no difference between the two experimental days regarding other adverse events (p > 0.05, McNemar’s test).
Migraine-like attacks and headache intensity after PGE2
Clinical characteristics of headache and associated symptoms in 12 migraineurs without aura during and after PGE2 and placebo (0–12 h).
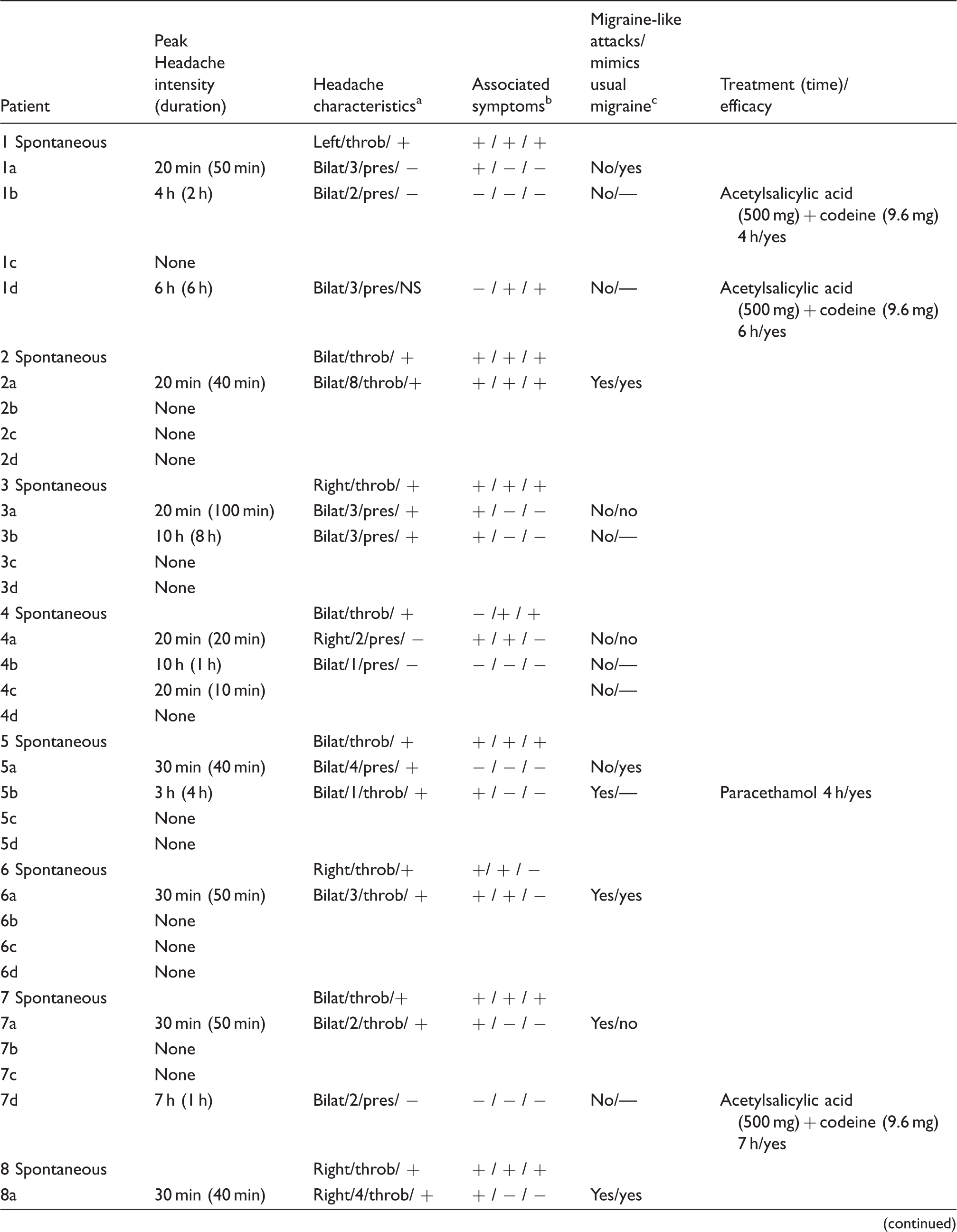

a: PGE2 day 0–90 min after infusion start; b: PGE2 day 1.5–2 h after infusion start; c: placebo day 0–90 min after infusion start; d: placebo day 1.5–12 h after infusion start; NS: not stated.
Localization/intensity/quality (throb: throbbing; pres: pressing)/aggravation (by cough in-hospital and by movement out-hospital phases).
Nausea/photophobia/phonophobia.
Migraine-like attacks are defined according to criteria, described in methods.
Incidence of headache intensity, nausea, photo- and phonophobia during the immediate phase for migraine patients and healthy subjects on PGE2 day.

Incidence of PGE2-induced headache and migraine-like attack in 12 migraine patients without aura.
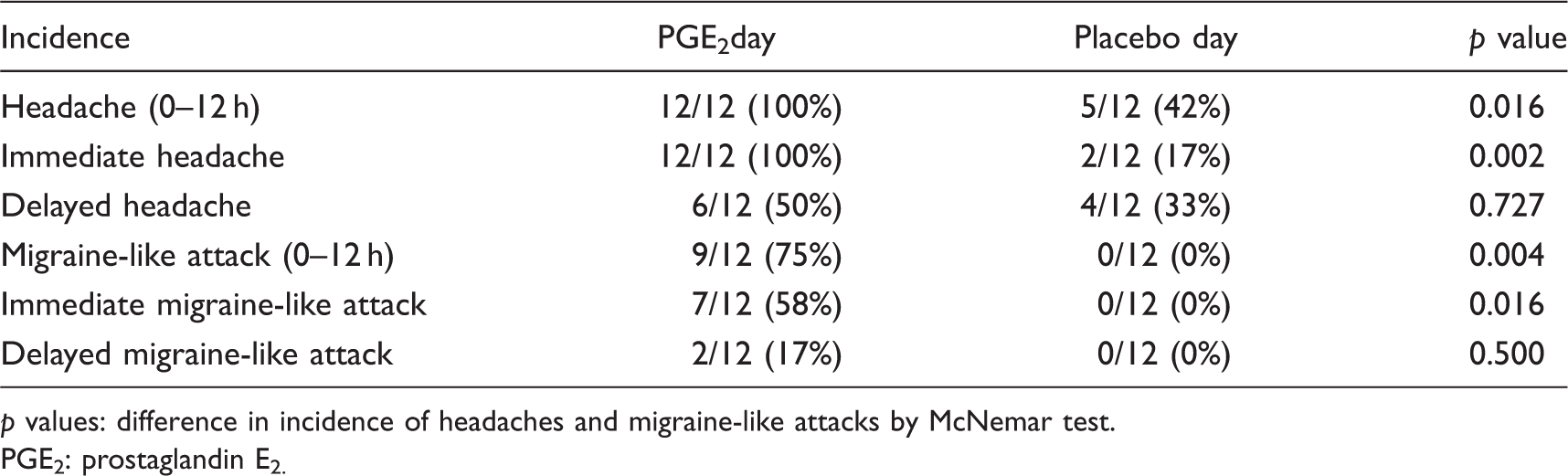
p values: difference in incidence of headaches and migraine-like attacks by McNemar test.
PGE2: prostaglandin E2.
The AUC for immediate headache (0–90 min) was significantly larger after PGE2, 8 (7–16.1), than after placebo, 0 (0–0) (p = 0.003). There was no difference in the AUC for delayed headache (1.5–12 h) between PGE2, 0.25 (0–3.8) and placebo, 0 (0–8.4) (p = 0.813). The median peak headache score was 3 (1–7) after PGE2 infusion and 0 (0–1) after placebo (Figure 1).
Median (filled shapes) and individual (open shapes) headache intensity on a verbal rating scale (VRS) on prostaglandin E2 (PGE2) and placebo days in 12 migraine patients without aura. The median peak immediate headache intensity after PGE2 was 3 (0–10 on VRS) and occurred at T30 after start of infusion. There was a significant difference in the area under the curve (AUC)0–90 min between PGE2 and placebo (p = 0.003). During out-hospital phase we found no difference in the AUC1.5–12 h between two experimental days (p = 0.813).
Vascular responses after PGE2
We found no difference in PetCO2 between two experimental days (p = 0.065). Since there were no differences in baseline VMCA between the left and the right side on either study day (p > 0.05), we used an average of both sides for further analysis. The AUCVMCA0–30 min was significantly different after PGE2 infusion than after placebo, mean difference (MD) 17.2 (confidence interval [CI]: 6.3–28, p = 0.005) (Figure 2A). The maximum percent change in diameter (Δd) of the MCA on PGE2 day was 7.2 ± 7.1% and occurred 10 minutes after the start of infusion.
Individual (open shapes) and mean (filled shapes) (A) flow velocities (cm/s) in the middle cerebral arteries (VMCA); (B) diameter (mm) of the superficial temporal artery (STA); (C) diameter (mm) of the radial artery (RA) after intravenous infusion of prostaglandin E2 (PGE2) and placebo in 12 migraine patients without aura. There was a significant difference in the area under the curve (AUC)VMCA 0–30 (p = 0.005) and the AUCRA (p = 0.0001), but there was no difference in the AUCSTA between PGE2 and placebo (p = 0.850).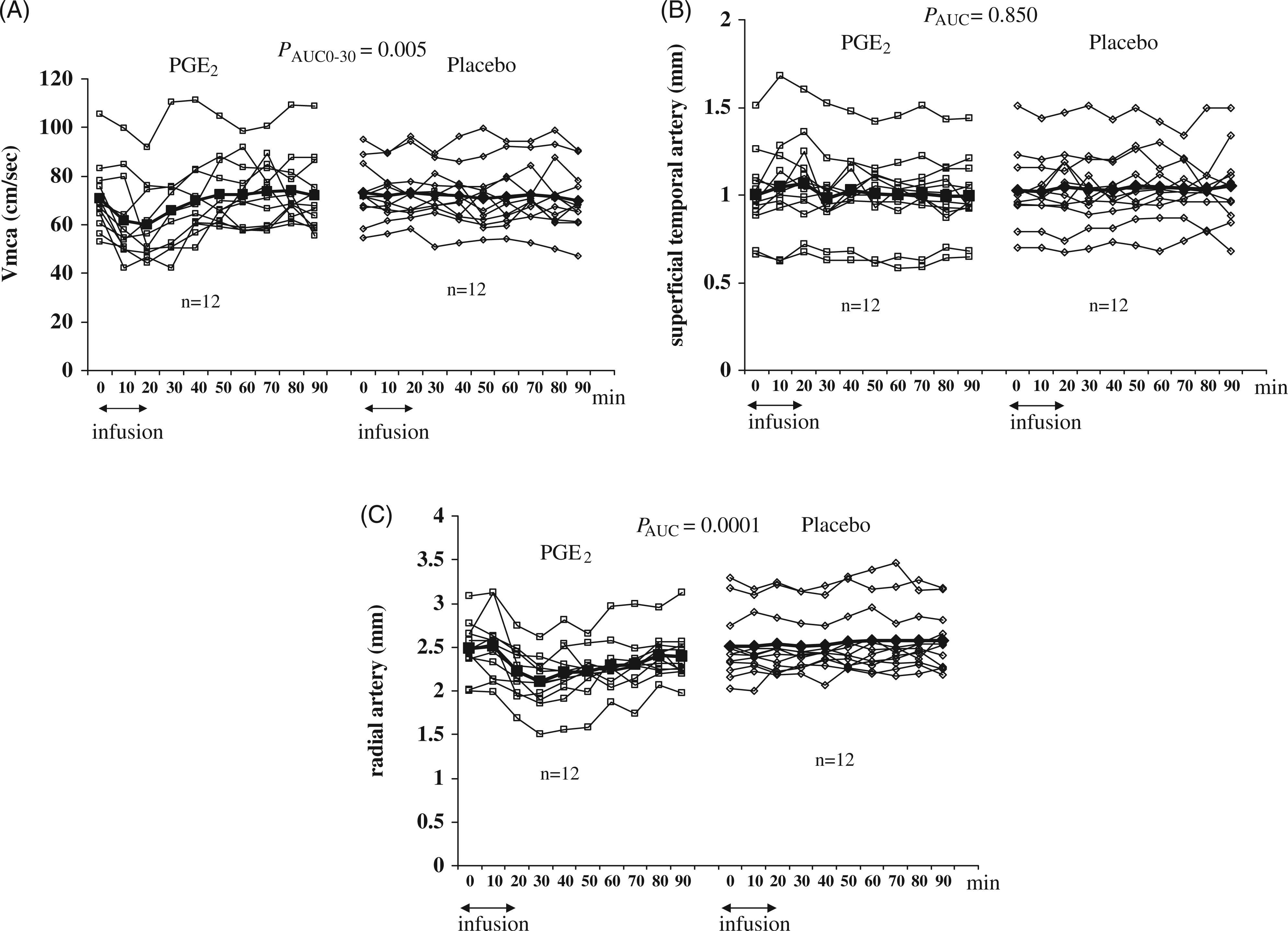
There was no difference in the AUCSTA between PGE2 and placebo, MD 0.04 (CI: −0.43–0.51, p = 0.850) (Figure 2B). A significant difference was found in the AUCRA between PGE2 and placebo, MD −1.9 (CI: −2.7– −1.1, p = 0.0001) (Figure 2C).
The AUC for HR was significantly larger after PGE2 than after placebo (p = 0.0001) (Figure 3). We found no difference in the AUCMAP between two experimental days (p = 0.482).
Individual (open shapes) and mean (filled shapes) changes in heart rate (HR) after intravenous infusion of prostaglandin E2 (PGE2) and placebo in 12 migraine patients. The area under the curve AUCHR was significantly larger on PGE2 day than on placebo day, mean difference 132 (confidence interval [CI]: 112–153, p = 0.0001).
Percentage changes from baseline of vascular parameters and median headache score are presented in Figure 4.
Percentage changes from baseline for mean values of blood flow velocity in the middle cerebral artery (VMCA), diameter of the superficial temporal artery (STA) and median headache score. VRS: verbal rating scale.
Discussion
The major outcome of the current study is that PGE2 was able to induce immediate migraine-like attacks in 58% of migraine patients. This is in contrast to previous pharmacological provocation experiments, where the majority of migraineurs reported delayed migraine-like attacks (15,19,22,23) (Figure 5). How can we explain the immediate migraine response after PGE2, which is in contrast to the previous studies? Could PGE2 be one of the important final products involved during migraine attacks? The puzzling difference we observed between PGE2 and PGI2 is interesting because both are products of the arachidonic acid cascade and one would expect similar migraine responses.
Percents of patients with immediate and delayed migraine-like attacks after glyceryltrinitrate (GTN), pituitary adenylate cyclise–activating peptide (PACAP), prostaglandin I2(PGI2), calcitonin gene–related peptide (CGRP) and prostaglandin E2 (PGE2) infusion.
We reviewed the previous experiments from our laboratory and found that only few patients (0–25%) reported immediate migraine-like attacks after pharmacological migraine triggers such as glyceryl trinitrate (GTN), pituitary adenylate cyclase–activating peptide (PACAP), prostaglandin I2 (PGI2) and calcitonin gene–related peptide (CGRP) (Figure 5). While there was no statistical difference in migraine-like attacks during the in-hospital immediate phase, the majority of patients (50–80%) reported delayed migraine-like attacks several hours after the infusion (15,19,22,23). The current provocation study using PGE2 revealed a clearly different response. Thus, 58% of patients (7 out of 12) reported immediate migraine-like attacks and only 17% (2 out of 12) reported delayed attacks. Furthermore, the following self-reported recordings suggest that patients experienced migraine-like attacks after PGE2 infusion: (1) 5 out of 7 (71%) had their usual (spontaneous migraine) localization of head pain; (2) 7 out of 7 (100%) had the same pain quality; (3) 6 out of 7 (86%) reported that the head pain and the associated symptoms were mimicking their spontaneous migraine attacks.
The present study showed that the vascular responses after infusion of PGE2 in migraineurs are in agreement with those that were found in the previous studies (15,19,22,23), apart from STA response. The similar non-significant response of STA compared to placebo we have previously reported in a provocation experiment after infusion of carbachol (24). Interestingly, both carbachol (25) and PGE2 (14) induced significant dilatation of STA in healthy volunteers, but not in migraineurs. We are unable to explain those findings. A difference between migraine patients and controls seems most likely in view of the high accuracy of measurement of STA (26). However, unexpectedly large intra-observer variability and a false-negative finding are also possible.
PGE2 and its receptor subtypes EP1–4 are present in both the peripheral (1,27–29) and central nervous systems (4,30,31). PGE2 plays a prominent role in both peripheral (32,33) and central sensitization (3,4,34–37). Intradermal injections of PGE2 in humans cause dose-related hyperalgesia (38). Application of inflammatory mediators on rat dura mater or electrical stimulation of trigeminal ganglion induced PGE2 release (8), most likely from branches of the middle meningeal artery, perivascular nerve endings and mast cells, where COX isoforms were found (39). Furthermore, an “inflammatory soup” used in animal experiments to sensitize trigeminal sensory afferents contains several inflammatory mediators, including PGE2 (40–42). The “inflammatory soup” – induced peripheral and central sensitization of trigeminal neurons is reversed by COX-inhibitors (41,42). Interestingly, trigeminal brain-stem neurones cannot be excited by the topical application of PGE2 alone on dura mater (43). However, its ability to sensitize meningeal nociceptors without the presence of the other mediators has previously not been studied.
The difference between the immediate migraine-like attack after PGE2 and the delayed migraine after PGI2 is interesting and unexpected. PGI2 sensitizes meningeal nociceptors (44) and hyperalgesia induced by PGI2 and PGE2 occur due to activation of EP1/IP–PKC cascade and activation of TRPV1 receptors of sensory neurones (45). Furthermore, PGI2 is considered to be a full agonist of EP1 and EP4 receptors (46). Therefore, it is likely that PGI2 might have induced immediate migraine-like attacks in the majority of migraineurs if given in a higher concentration. It was not possible to increase the PGI2 concentration due to a risk of major blood pressure decrease accompanied by HR increase over the safety limits, defined in the protocol (15). In contrast to PGI2, the absence of the immediate migraine-like attacks during glyceryltrinitrate (GTN), CGRP and PACAP infusions is more expected. Nitric oxide (NO) can both sensitize and inhibit mechanosensitive dural nociceptors (47). The increase of c-Fos expression in the trigeminal ganglion (TG) and trigeminal nucleus caudalis (TNC) was observed four hours after the intraperitoneal injection of either GTN or PACAP in mice (48). CGRP does not sensitize meningeal nociceptors (49). Interestingly, NO stimulates COX synthesis and PG production (50). Intraperitoneal GTN administration in rats caused increase of COX-2 followed by an PGE2 increase in the hypothalamus and lower brain stem (51). CGRP has also been shown to induce secondary liberation of PGE2 (52). Moreover, IL-1beta stimulates COX-2-dependent PGE2 synthesis and CGRP release in rat trigeminal ganglia (53). Based on these data, it would be plausible to suggest that PGE2 is one of the final products involved in spontaneous and experimentally induced migraine attacks.
Given that sensitization of the sensory neurones by PGE2 is mediated through EP1–4 receptor subtypes, (54,55) these receptors could be considered as a new potential drug targets. We have previously shown that a specific EP4 receptor antagonist is not able to prevent PGE2-induced headache in healthy volunteers (56). Hyperalgesia induced via EP2 and EP3 receptors cannot be abolished by EP1 receptor antagonist alone (57). Trigeminal nociceptors express EP2 and EP3 receptors co-expressed with TRPV1 receptors (58). Blockade of a single receptor is probably not sufficient to reverse PGE2-induced sensitization. It would, therefore, be interesting to investigate if a combined EP2 and EP3 receptor antagonist could prevent PGE2-induced migraine-like attacks and thereby become a possible new anti-migraine drug target.
Footnotes
Funding
The study was sponsored by IMK Almene Fond and the Danish Headache Society.
Acknowledgements
We thank Lene Elkjær (LE) and Winnie Grønning (WG) for excellent technical support.
Conflict of interest/disclosure statement
The authors were independently responsible for the study design, data analysis and manuscript. The authors report no conflict of interest in relation to this study. The study was sponsored by IMK Almene Fund.