
Abstract
Introduction: In recent years the kynurenine family of compounds, metabolites of tryptophan, has become an area of intensive research because of its neuroactive properties. Two metabolites of this family have become of interest in relation to migraine and pain processing.
Discussion: Experimental studies have shown that kynurenic acid (KYNA) plays an important role in the transmission of sensory impulses in the trigeminovascular system and that increased levels of KYNA decrease the sensitivity of the cerebral cortex to cortical spreading depression. Furthermore, another metabolite of the kynurenine family, L-kynurenine, exerts vasodilating effects similar to nitric oxide by increasing cyclic guanosine monophosphate.
Conclusion: This review summarizes current knowledge of the role of kynurenine signalling in trigeminal and central pain processing, including its therapeutic prospects in migraine treatment.
Introduction
The kynurenine family of compounds (kynurenines) are metabolites of the essential amino acid tryptophan (1). Kynurenines consist of L-kynurenine (L-KYN), kynurenic acid (KYNA), quinolinic acid, 3-hydroxykynurenine and anthranilic acid. In 1981, Stone and Perkins (2) reported that quinolinic acid may activate the subpopulation of neuronal glutamate receptors sensitive to N-methyl-D-aspartate (NMDA). Since then, around 4000 papers have been published on the intermediates of the kynurenine pathway and in particular on the role of KYNA because of its neuroprotective effects by inhibiting glutamate release and neuronal transmission (3). In recent years there has been increasing interest in the role of KYNA in trigeminal pain processing (4–7). Given the important role of the trigeminovascular system (8,9) and glutamate (10,11) in the pathophysiology of migraine, it would be plausible to suggest that the kynurenine pathway (see below, and Figure 1) (12) may be a new target for anti-migraine drugs. This review focuses primarily on the role of KYNA in nociception and in particular on the most recent findings in relation to trigeminal pain processing, and its therapeutic prospects in migraine treatment.
The kynurenine pathway. Reproduced by permission from reference (12).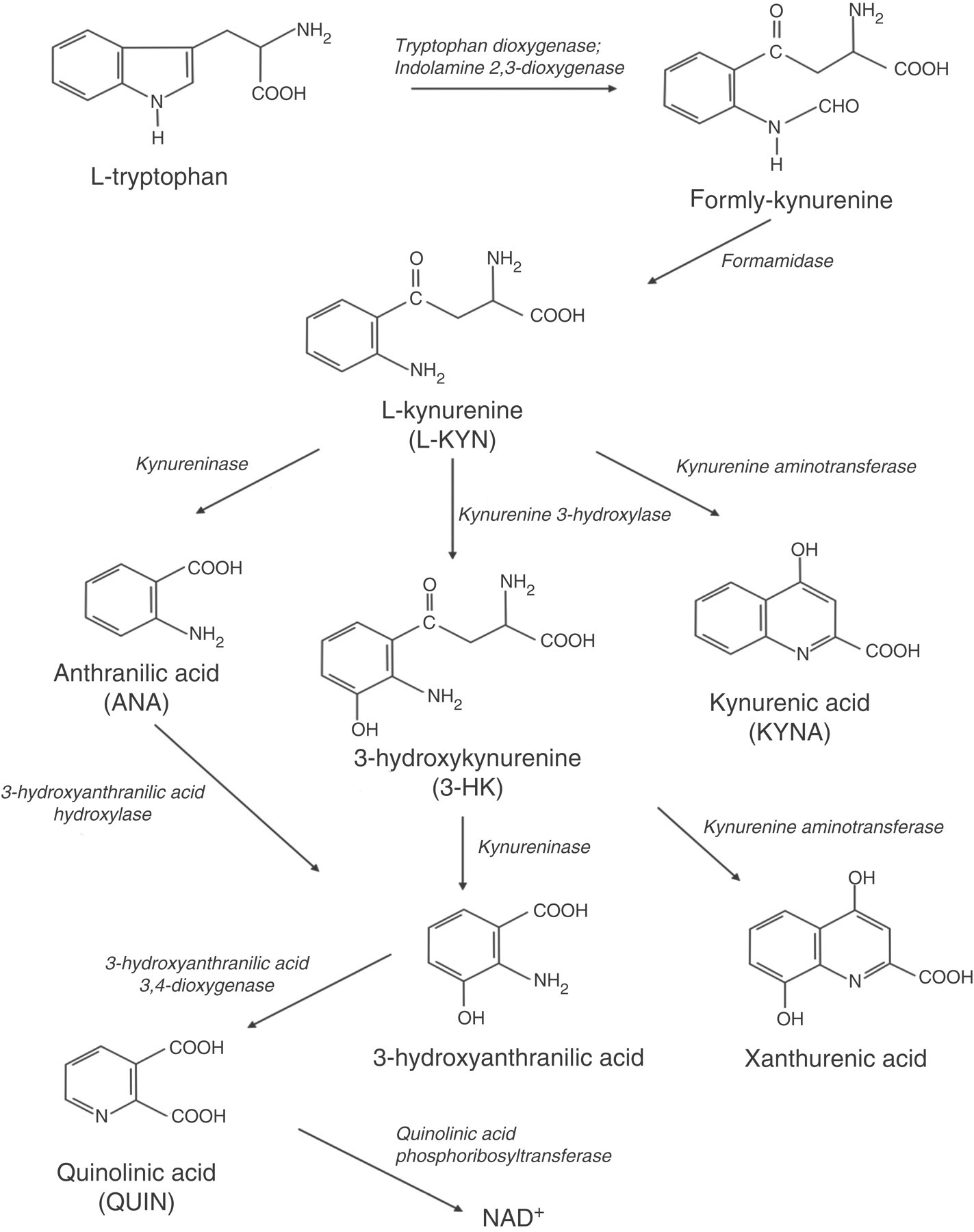
History
L-KYN has been recognized as a product of tryptophan since 1931 (13), and in 1947 the kynurenine pathway was recognized as a major route of tryptophan metabolism (1). The whole kynurenine pathway and its metabolites were mapped for the first time in 1969 (14). In 1981, Stone and Perkins (2) reported that quinolinic acid might activate NMDA-receptors and affect neuronal transmission. A year later the neuro-inhibitory activities of KYNA were reported (3), and KYNA became an important focus of neurological research.
The kynurenine pathway
L-KYN is a metabolite of the amino acid tryptophan. The tryptophan metabolic pathway leading to the synthesis of kynurenine (Figure 1) is the main route for non-protein metabolism of the essential amino acid tryptophan with nicotine adenine dinucleotide (NAD+) and NAD-phosphate (NADP+) as end products (15). About 80% of tryptophan metabolism is metabolized through the kynurenine pathway by the kynurenine aminotransferases (KATs) (16). Another important pathway of tryptophan metabolism is the serotonin pathway.
The first step of the kynurenine pathway is engaged by two enzymes: tryptophan-2,3-dioxygenase and indoleamine-2,3-dioxygenase (IDO), which differ in their tissue localization and regulation. Tryptophan-2,3-dioxygenase is predominantly expressed in the hepatic cells regulated by glucocorticoids (17), while IDO is distributed in peripheral tissues (lung, spleen, etc.) and the nervous system (neurons, glial cells, astrocytes) (18–20) activated by proinflammatory cytokines (21–23).
L-KYN is converted from tryptophan by tryptophan-2,3-dioxygenase/IDO and is the source for the synthesis of all the other metabolites (kynurenines) of the kynurenine pathway. In the brain, L-KYN can be converted to four metabolites: KYNA, quinolinic acid, 3-hydroxykynurenine and anthranilic acid. The synthesis of KYNA from L-KYN is catalysed in an irreversible process by L-KYN aminotransferase, while quinolinic acid is produced indirectly from L-KYN catalysed by 3-hydroxy-ANA oxygenase in the last step of its synthesis (Figure 1). There are two types of L-KYN aminotransferases in the nervous system: KAT I and KAT II. KAT II is the one primarily responsible for the formation of KYNA in the brain (24).
There are several enzymes that take part in the cascade. Investigation of these enzymes may be of interest, because their modification could shift L-KYN metabolism towards the excitotoxin quinolinic acid or the neuroprotective KYNA, but so far no consistent patterns have emerged to identify specific functions of the cerebral kynurenine pathway.
Distribution in the peripheral nervous system and central nervous system
L-KYN and its derivative KYNA are found in almost all organs and tissues of the human body because of the wide distribution of IDO (25), the crucial intracellular enzyme that engages the formation of L-KYN. At present there are, to our knowledge, no reports on L-KYN’s distribution in the peripheral nervous system (PNS) including the dorsal root ganglion or trigeminal ganglion. Approximately 40% of the L-KYN in the brain is synthesized there; the remainder is taken up from the periphery (60%). This indicates that L-KYN can be easily transported across the blood–brain barrier by the neutral amino acid carrier (26). In contrast to L-KYN, KYNA has only a very limited ability to cross the blood–brain barrier (26). However, experimental data suggest that peripheral treatment with L-KYN dose-dependently increases the concentration of KYNA in the brain (27). IDO has been shown to be distributed evenly throughout the brain (28). It is not normally expressed in cells but requires induction by IFN-γ, tumour necrosis factor or lipopolysaccharides (22,23,29). During systemic inflammation, vascular endothelial cells are the primary site for IDO expression in vivo and induce formation of L-KYN (30). However, L-KYN can still be found widely in the brain under non-inflammatory conditions (25).
In the CNS, the metabolites of the kynurenine pathway, including L-KYN and KYNA, have been detected in macrophages, microglia cells, astrocytes and neurons (18–20). All the enzymes of the pathway are primarily contained in astrocytes and microglia cells (18,19,24). Interestingly, astrocytes seem not to contain kynurenine 3-hydroxylase and therefore favour KYNA synthesis (Figure 1). In contrast, microglia cells have very little kynurenine aminotransferase activity (Figure 1) and therefore primarily form intermediates of the quinolinic acid branch of the pathway (20).
In summary, KYNA is produced primarily by astrocytes and neurons in the CNS. Given that kynurenine metabolites are widely distributed in the brain, it is likely that KYNA is present in structures relevant for migraine such as the trigeminal nucleus caudalis, periaqueductal grey, nucleus raphe, thalamus and hypothalamus.
Receptors
L-KYN has been suggested to be an endothelium-derived vasodilator by activating the cGMP-dependent pathway, because of its ability to activate purified rat soluble guanylate cyclase and to increase tissue concentrations of cGMP (31). Furthermore, data suggest that L-KYN also activates the cAMP pathway, as shown by its ability to increase the activity of adenylate cyclase and increase the cAMP content in porcine coronary arteries and in rat aortic smooth muscle cells (31,32). At present, it is unknown whether L-KYN per se exerts neuroprotective or neurotoxic effects in the nervous system like its metabolites, which are known to exert notable neuroactive properties on the glutamate receptors, in particular on the NMDA receptors and alpha-amino-3-hydroxy-5-methyl-4-isoxazole propionic acid (AMPA) receptors (33).
The NMDA receptor is one of the major classes of ionotropic excitatory amino acid receptors. NMDA receptors require co-activation by two ligands, glutamate and glycine, and are known to play a crucial role in the development or maintenance of central sensitization, i.e. increased excitability of neurons in the CNS (34–37). KYNA is a neuroprotective agent, which acts on three different receptors: the excitatory amino acid receptors (NMDA/AMPA), α7 nicotinic acetylcholine receptors and GPR35 receptors. KYNA is the only known endogenous NMDA receptor antagonist, which in particular acts at the strychnine-insensitive glycine binding site of the NMDA receptor (Figure 2) (38). It also exerts a weak antagonistic effect on the AMPA receptors (39). Experiments have shown that KYNA is a janus-faced compound that exerts different effects on the NMDA and AMPA receptors in high (micromolar) and low (nanomolar) concentrations. Thus, in vitro electrophysiological examinations on young rat hippocampus confirmed that KYNA in micromolar concentrations exerts an inhibitory effect (39,40). However, in nanomolar concentrations, KYNA does not give rise to inhibition, but in fact facilitates the field excitatory post-synaptic potentials. Based on these, it has been suggested that KYNA in the concentration range between a few hundred nanomolar and micromolar displays different effects (41).
Effect of kynurenic acid (KYNA) on the glutamatergic system through NMDAR and AMPAR. Reproduced with permission of the Massachusetts Medical Society. Source: Katzung BG, Masters SB, Trevor AJ. Basic and Clinical Pharmacology, 11th edn. http: //www.accessmedicine.com) MGluR, metabotropic glutamate receptor; AMPAR, alpha-amino-3-hydroxy-5-methyl-4-isoxazole propionic acid receptor; NMDAR, N-methyl-D-aspartate receptor.
KYNA also acts as a non-competitive blocker of the α7-nicotinic acetylcholine (α7-nACh) receptors (42). Data indicate that KYNA significantly decreases extracellular glutamate levels in rat brain through its antagonist activity on α7-nACh receptors (43) and contributes to the inhibitory effects of KYNA.
Recent studies have shown that KYNA is a ligand for the orphan G-protein-coupled receptor GPR35, evoking a specific rise in [Ca2+] i in cells expressing human GPR35 (44), which is expressed by nociceptive neurons in the dorsal root ganglion (45). KYNA can inhibit the over-excitation of glutamatergic transmission by modulating the activity of NMDA receptors and α7-nACh receptors. Interestingly, KYNA might have a role in stimulating the peripheral glutamate receptors as well (46).
In summary, L-KYN per se does not seem to have any direct neuroactive properties. It increases cAMP and cGMP through activation of adenylate cyclase and soluble guanylate cyclase in arteries. On the other hand, L-KYN may modulate neuro-inhibitory activities indirectly through its derivative KYNA, which affects the NMDA receptors and modulates glutamate levels in the CNS.
Peripheral and central pain processing
A growing body of evidence indicates that KYNA has a mediatory and modulatory role for glutamate and its receptors in peripheral and central pain processing (25,37,47,48). A recent study has demonstrated that KYNA inhibited forskolin-stimulated cAMP production by cultured rat dorsal root ganglion neurons (45). It has been reported that KYNA exerts an anti-nociceptive effect by inactivating peripheral glutamate receptors and attenuating sensitization of cutaneous nociceptors (49,50). Furthermore, intraperitoneal administration of KYNA decreases the nociceptive behaviour in both the tail flick and hot plate tests (51). Injection of glutamate into the rat masseter muscle in vivo, which increased muscle blood flow by 250%, can be attenuated by KYNA (52). Given that injection of glutamate into the human masseter muscle evokes pain with increased blood flow (53), it is possible that KYNA may have peripheral analgesic effects mediated through inhibition of peripheral glutamate receptors.
Regarding the CNS, iontophoretic administration of KYNA to the spinal cord can markedly reduce the nociceptor response of the spinal cord neurons after cutaneous and muscular noxious stimulation (48). The nociceptive response was found by measuring the activity of the wide dynamic range neurons in laminae IV–VI of the dorsal horn and the results suggest that NMDA receptors preferentially mediate transmission of nociceptive information originating in skin and muscle. Furthermore, intracisternal injection of KYNA may attenuate capsaicin and formalin-induced nociception in the rat (37,54). In addition, noxious activation of the locus coeruleus neurons is blocked by intracerebroventricular injection of KYNA (34). The increase in KYNA in the locus coeruleus was obtained by a potent inhibitor of kynurenine 3-hydroxylase, which is able to dose-dependently increase the levels of KYNA in the brain. This treatment abolished the increase in firing rate of locus coeruleus neurons. Another study (55) examined how blockade of the glutamatergic system by injection of L-KYN leading to a rise of KYNA within the periaqueductal grey modifies the projection pathway of medial pre-optic nucleus of the hypothalamus to nucleus raphe magnus. It showed that the periaqueductal grey cell activity can be inhibited significantly by the injection of L-KYN in the periaqueductal grey and modulates the neural transmission between the structures involved (55). Moreover, glutamatergic stimulation of the nucleus raphe magnus could be abolished by KYNA administration (56).
Collectively, these data show that KYNA has an inhibitory effect and can modulate neuronal transmission in both the PNS and CNS.
The kynurenine pathway in trigeminal pain processing
Kynurenines may exert inhibitory and excitatory actions on both pre- and post-synaptic sites in the trigeminovascular system (Figure 3). It has been shown that Schwann cell membranes surrounding nerve fibres in the supratentorial dura mater display kynurenine aminotransferase-immunoreaction (57). Interestingly, stimulation of the trigeminal ganglion decreased the kynurenine aminotransferase immunoreaction considerably, probably due to decrease in the concentration of KYNA (57). At the same time, the Schwann cells in the dura mater became conspicuously swollen, while nitric oxide synthase of nerve fibres in the dura mater increased, suggesting release of nitric oxide. The latter is known to be involved in NMDA receptor activation leading to vasodilatation followed by neurogenic inflammation (58).
The effects of kynurenine metabolites in the trigeminovascular system. Modified by permission from reference (90). From Goadsby PJ, Lipton RB and Ferrari MD. Migraine current understanding and treatment. N Engl J Med 2002; 346: 257–270. Reproduced with the permission of McGraw-Hill Education.
Recent studies have suggested that KYNA plays an important role in the transmission of impulses in the trigeminal nucleus caudalis from the central terminals of the first-order to second-order trigeminal neurons, the latter projecting to the thalamus (4–7). A rat model of trigeminovascular activation by electrical stimulation has shown that L-KYN, in combination with probenicid, is able to reduce c-fos activation in the trigeminal nucleus caudalis and to increase KYNA levels in the nucleus (7). Probenicid aids the transport of L-KYN over the blood–brain barrier, and inhibits the transport of organic acid from the cerebrospinal fluid, thereby increasing the concentration of KYNA in the brain (4). Similar studies have also reported that KYNA could attenuate the nitroglycerin-induced neuronal nitric oxide synthase, calmodulin-dependent protein kinase II alpha (CamKIIa) and CGRP immune-reactivity in the rat trigeminal nucleus caudalis (6). The most likely explanation is that KYNA blocked the activation of first-order neurons and the consecutive release of CGRP and nitric oxide from the nerve endings (6). In the rat, nitroglycerin activates second-order neurons in the trigeminal nucleus caudalis, and increases the expression of neuronal nitric oxide synthase and CamKIIa, and induces CGRP release (5,59).
In addition to the peripheral effects, KYNA may also act at the central level. Intrathecal injection of KYNA produces dose-dependent and reversible analgesic effects in the hot plate, tail flick and formalin tests of nociception in mice and in rats (37,60,61). Furthermore, KYNA exerts a modulating effect by inhibiting the neural activity on brain stem structures such as the locus coeruleus, periaqueductal grey and nucleus raphe magnus (34,55,56,62).
Cortical spreading depression (CSD), an event believed to underlie visual aura in migraine, may activate and sensitize trigeminal nociceptors in animal models of migraine (63–65). CSD increases the level of KYNA in the rat cortex (66). The increase of KYNA after CSD is possibly a neuroprotective response. Injection of probenicid probably produces a much greater increase in KYNA concentration in the CNS. Moreover, administration of probenicid via microdialysis showed a marked decrease in CSD in the rat striatum, possibly by increasing the brain KYNA concentration (67).
Taken together, these data suggest that KYNA may play an important role in trigeminal pain processing.
A potential therapeutic target in migraine?
The pathophysiological mechanisms underlying migraine pain include abnormal processing in trigeminal nociceptors in the dura mater and deep brain structures including the trigeminal nucleus caudalis, periaqueductal grey and thalamus (68,69). Provocation studies in migraine sufferers using intravenous administration of vasoactive substances such as the nitric oxide donor glyceryl trinitrate and CGRP have identified two major signalling pathways in migraine pathogenesis: nitric oxide-cGMP (70,71) and CGRP-cAMP (72,73). In addition, 5-HT1B/1D agonists still represent the gold standard for the treatment of migraine attacks. The question is whether the kynurenine pathway interacts with cGMP, cAMP and serotonin signalling relevant for migraine and whether the modulation of kynurenine signalling may provide a novel target for migraine prevention.
Recent data indicate that L-KYN has vasodilating effects like nitric oxide, and also activates the cGMP-dependent pathway (74). Intravenous infusion of L-KYN decreases mean arterial blood pressure of hypertensive rats in a dose-dependent manner and relaxes constricted arteries (31). L-KYN increases both cGMP and cAMP concentrations during inflammation, because of induced activation of IDO (Figure 4) (31). L-KYN stimulates the activities of soluble guanylyl cyclase and adenylyl cyclase in smooth muscle cells from porcine coronary arteries (31). The finding that L-KYN stimulates mainly the haem-free form of soluble guanylyl cyclase is interesting, because normally nitric oxide leads to an increase in soluble guanylyl cyclase activity which, in turn, catalyses the conversion of guanosine triphosphate to cGMP and pyrophosphate (32). However under inflammatory conditions, soluble guanylyl cyclase is oxidized to become a haem-free enzyme that is refractory to activation by nitric oxide (31,32). Thus, L-KYN can stimulate this inactive soluble guanylyl cyclase to produce cGMP. Furthermore, intravenous infusion of L-tryptophan dilates coronary arteries and produces an increase in the regional cerebral blood flow (75). However the dilating effect of tryptophan requires the presence of active IDO and an intact endothelium, whereas the effect of kynurenine was endothelium-independent (31). These findings suggest that L-KYN might be the vasoactive metabolite of tryptophan, but the study cannot exclude the possibility that a kynurenine derivative present in the preparation of kynurenine was in fact responsible for some of the activities observed. Nonetheless, the kynurenine is able to activate the cGMP and cAMP pathways, which are important signalling pathways in migraine.
L-KYN and nitric oxide (NO) regulation of cAMP and cGMP pathways in vascular smooth muscle tone under inflammation. Arg, arginine; Trp, tryptophan; sGC, soluble guanylate cyclise; sGC-Hf, soluble guanylate cyclase-haem-form; AC, adenylate cyclase.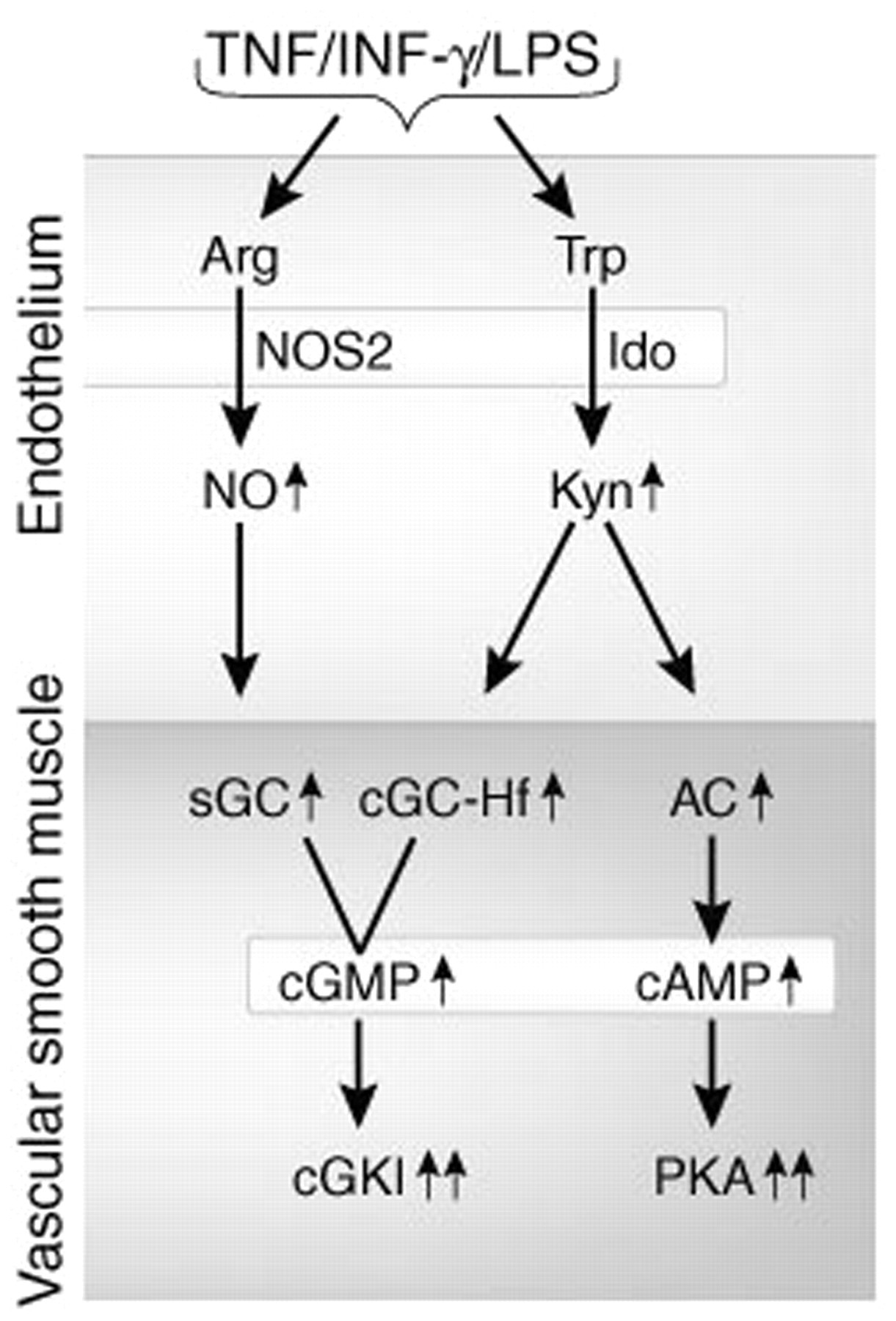
The formation of the L-KYN seems directly proportional to tryptophan concentrations (76). Tryptophan has been used both experimentally and therapeutically as a mean of increasing tryptophan conversion to serotonin (77). Interestingly, it has been shown that the activation of IDO shifts tryptophan metabolism from serotonin synthesis to formation of the kynurenines (78). The formation of L-KYN, which competes for cerebral transport and cellular uptake with L-tryptophan, may actually decrease the production of cerebral serotonin (76). Low cerebral serotonin levels have been implicated in the pathophysiology of migraine (79,80).
Based on these data it would be plausible to suggest that L-KYN may have a notable role in the pathogenesis of migraine because of its vasodilating properties similar to nitric oxide, by activating the cGMP pathway and its presumed ability to affect the cAMP and serotonin pathways as well. Development of drugs that inhibit the effect of IDO or L-KYN on soluble guanylyl cyclase or serotonin pathways might therefore be a target of further migraine research and drug development.
Another way to modulate the kynurenine pathway is to raise the concentration of the neuroprotective KYNA in the brain. As an endogenous NMDA receptor antagonist and acting at the strychnine-insensitive glycine binding (38), KYNA exerts an antinociceptive action in the trigeminovascular system (81,82). Furthermore, increased levels of KYNA decrease the sensitivity of the cerebral cortex to CSD (83). These data suggest that KYNA could be a possible candidate for the treatment of migraine with and without aura.
Based on experimental data of the kynurenine pathway we suggest two possible ways in which therapeutic agents could be developed for the treatment of migraine. One approach is to fend off the effects of a kynurenine pathway disturbance using enzyme inhibitors, which can shift the kynurenine pathway towards the neuroprotective KYNA and inhibit the accumulation of quinolinic acid and other neurotoxic metabolites (12,15,84). The second approach is to use analogues of KYNA as antagonists at the ionotropic glutamate receptors (85). KYNA may, as mentioned earlier, pass the blood–brain barrier poorly, and it would be important to develop different analogues of KYNA that can cross the blood–brain barrier easily and display similar effectiveness on the affected receptors at specific locations in the brain (86). In support, a study showed that systemic treatment with SZR72, a synthetic NMDA receptor antagonist KYNA analogue, abolished nitroglycerin-induced c-fos activation in the trigeminal nucleus caudalis (87). Blocking vasoactive effects of L-KYN might also be interesting, but no studies have apparently been done.
Conclusion
Growing evidence suggests that the metabolites of the kynurenine pathway may play a crucial role in the modulation of pain processing both peripherally and centrally. Animal studies have shown that elevating levels of KYNA by inhibiting the glycine co-agonist site of the NMDA receptors in the brain may modify or inhibit nociceptive transmission and reduce extracellular glutamate. A proof of concept study on the effect of a metabotropic glutamate receptor 5 (MGluR5) antagonist in the treatment of migraine attacks (88) supports the involvement of glutamate and NMDA receptors in migraine pathogenesis. MGluR5 increases NMDA receptor activity, thus effects on this receptor alter the activity of the ionotropic receptors (89). KYNA is involved in sensory processing in the PNS and CNS, including in migraine-relevant structures such as the trigeminal nucleus caudalis, periaqueductal grey and locus coerulus. L-KYN exerts vasodilating effects similar to nitric oxide by increasing cGMP, which is an integral part of migraine pathogenesis. Furthermore, L-KYN also modulates the cAMP and serotonin pathways. Collectively, drugs interacting on the kynurenine pathway may provide a novel target for migraine prevention.
Footnotes
Funding
This work was supported by The Lundbeck Foundation Center for Neurovascular Signaling (LUCENS).
Conflict of interest
None in relation to this paper.
Acknowledgements
The authors thank Professor Jes Olesen for reading the manuscript and giving valuable comments.