
Abstract
Objective: Spontaneous intracranial hypotension (SIH) is caused by spinal cerebrospinal fluid (CSF) leakage. An underlying connective tissue disorder has been hypothesized to cause dural weakness and predisposition to CSF leak. We conducted a case-controlled study to investigate the role of connective tissue disorders in SIH patients.
Methods: We recruited 55 consecutive SIH patients (38 F, 17 M; mean age, 40.8 ± 9.8 years) and 55 age- and sex-matched control individuals (mean age, 38.0 ± 8.9 years) for this study. The connective tissue disorders were evaluated by: (i) Beighton hypermobility scores and revised diagnostic criteria for benign joint hypermobility syndrome; (ii) skin and skeletal manifestations of Ehlers–Danlos syndrome (EDS); and (iii) skeletal features of Marfan syndrome.
Results: The frequencies of joint hypermobility according to Beighton scores >4/9 (SIH 23.6% vs controls 16.4%, P = 0.48) and revised benign joint hypermobility syndrome criteria (SIH 23.6% vs controls 34.5%, P = 0.29) did not differ between SIH patients and controls. Sixteen patients and 16 controls had one or more skin features of EDS (P = 1.0). Nine SIH patients (16.4%) demonstrated the skeletal features of Marfan syndrome; this frequency did not differ from that of the control group (9.1%; P = 0.262). Only dolichostenomelia (disproportionately long limbs) was more prominent in SIH patients than in controls (34.5% vs 9.1%; P = 0.002).
Conclusion: Compared with Western studies, the frequencies of connective tissue disorders were higher in our SIH patients. However, these frequencies did not differ between SIH patients and control individuals, except for dolichostenomelia.
Keywords
Introduction
Spontaneous intracranial hypotension (SIH) is characterized by orthostatic headaches that are relieved by recumbency. This disorder is caused by the leakage of spinal cerebrospinal fluid (CSF) without a preceding dural puncture or other procedure or trauma that may cause a CSF fistula (1). The exact cause of spontaneous spinal CSF leakage remains unknown, but the existence of an underlying connective tissue disorder has been hypothesized as contributory to dural weakness in patients with SIH (2). Two studies (3,4) in US populations found that an estimated 16–38% of SIH patients had connective tissue disorders, including joint hypermobility, Ehler–Danlos syndrome (EDS), and Marfan syndrome.
The prevalence of joint hypermobility varies according to age, sex, and race. Beighton hypermobility scores have indicated that 17% of Asian populations (5) and 5% of Western populations (6) have this disorder. The association of connective tissue disorders with SIH in Asian populations, however, is unknown. Furthermore, although previous studies (3,4) have associated SIH with connective tissue disorders, they did not recruit healthy control individuals for comparison.
In this study, we evaluated the underlying connective tissue disorder profiles of SIH patients, and compared these data to those of corresponding healthy control individuals.
Subjects and Methods
For this study, we prospectively enrolled consecutive SIH patients at Taipei Veterans General Hospital from January 2007 to November 2009. Age- and sex-matched control subjects were recruited, mainly from the hospital staff. SIH was diagnosed based on the criteria provided by the second edition of the International Classification of Headache Disorders (ICHD-2) (7):
Orthostatic headache associated with at least one of the following: neck stiffness, tinnitus, hypacusia, photophobia, and nausea. At least one of the following: (i) evidence of low CSF pressure on magnetic resonance imaging (MRI); (ii) evidence of CSF leakage on conventional myelography, computed tomographic myelography, or cisternography; and (iii) CSF opening pressure less than 60 mmH2O. No history of dural puncture or other causes of CSF fistula.
ICHD-2 criterion 4, headache resolves within 72 h after epidural blood patches, was not used in this study because not all patients underwent this procedure.
One author (FC Liu) performed the physical examinations and took the subject histories of connective tissue disorder features for all SIH patients and control subjects. The examinations and histories focused on benign joint hypermobility syndrome, EDS, and Marfan syndrome. The study was approved by the Institutional Review Board of Taipei Veterans General Hospital, and the patients gave informed consent.
The diagnosis of benign joint hypermobility syndrome was based on the Beighton hypermobility score and the revised (Brighton 1998) criteria for benign joint hypermobility syndrome (8). The Beighton hypermobility score evaluates the following: (i) passive dorsiflexion of the fifth digit beyond 90°; (ii) passive apposition of the thumbs to the flexor aspect of the forearms; (iii) hyperextension of the elbows; (iv) hyperextension of the knees; and, lastly, (v) forward flexion of the trunk with knees fully extended so that the palms of the hands rest flat on the floor. One point is given for manoeuvres (i–iv) on each side and one point is given for manoeuvre (v), for a maximum total score of nine points. A total score greater than four designates joint hypermobility.
The revised criteria for benign joint hypermobility syndrome base a diagnosis on the presence of two of the following major criteria, one major and two minor criteria, or four minor criteria. The major criteria are a Beighton score of four or greater, and arthralgia for longer than 3 months in four or more joints. The minor criteria are: (i) a Beighton score of 1–3 (0–3 if age >50 years); (ii) arthralgia for at least 3 months in 1–3 joints; (iii) back pain for at least 3 months; (iv) spondylosis, or spondylolysis and/or spondylolisthesis; (v) dislocation/subluxation in more than one joint, or in one joint on more than one occasion; (vi) soft-tissue rheumatism with at least three lesions; (vii) Marfanoid habitus; (viii) abnormal skin characterized by striae, hyperextensibility, thinness, or papyraceous scarring; (ix) eye signs such as drooping eyelids, myopia, or antimongoloid slant; and (x) varicose veins, hernia, or uterine/rectal prolapse.
The clinical features and diagnosis of EDS were based on the 1997 revised nosology of Villefranche (9). The skin manifestations of EDS include hyperextensibility, smooth and velvety texture, extensive bruising, thinness and translucency, and atrophic scarring. Extensive bruising data were obtained from the subject histories and the other characteristics were evaluated during objective physical examination.
The hypermobility type of EDS was diagnosed if two of the following major criteria and two minor criteria were present. The major criteria were generalized joint hypermobility indicated by a Beighton score of five or greater, and skin involvement (hyperextensibility and/or smooth, velvety skin); and the minor criteria were recurring joint dislocations, chronic musculoskeletal pain, and a positive family history (10).
The skeletal features of Marfan syndrome consist of pectus carinatum, pectus excavatum; (body-from-hips-to-top-of-head) as a ratio to (body-from-hips-to-feet) less than 0.89, arm-span-to-height ratio greater than 1.05; presence of wrist sign (thumb overlapping the terminal phalanx of the fifth digit when grasping the contralateral wrist) or thumb sign (entire thumbnail projecting beyond the ulnar border of the hand when the hand is clenched without assistance); reduced extension at the elbows; and scoliosis. The presence of at least two of these components indicates the involvement of the skeletal system in Marfan syndrome (11). Medial displacement of the medial malleolus causing pes planus and protrusio acetabuli were not evaluated in this study because radiological studies were not performed.
Statistical analysis
All statistical analyses were performed using SPSS v15.0 for Windows (SPSS Inc., Chicago, IL, USA). For continuous measures, Student’s t-test was used to examine the difference between patient and control groups. For categorical data, chi-squared or Fisher’s exact tests were used. All P-values were two-tailed and statistical significance was defined as a P-value of less than 0.05.
Results
During the study period, 55 SIH patients (38 females, 17 males; mean age, 40.8 ± 9.8 years; range, 24–67 years) were recruited. The control group also consisted of 55 subjects (38 females, 17 males; mean age, 38.0 ± 8.9 years; range, 24–62 years). The mean body height (164.0 cm vs 163.1 cm; P = 0.61) and weight (61.2 kg vs 61.8 kg; P = 0.83) did not differ between patient and control groups.
Comparisons of the frequencies of connective tissue disorders in patients with spontaneous intracranial hypotension and control subjects
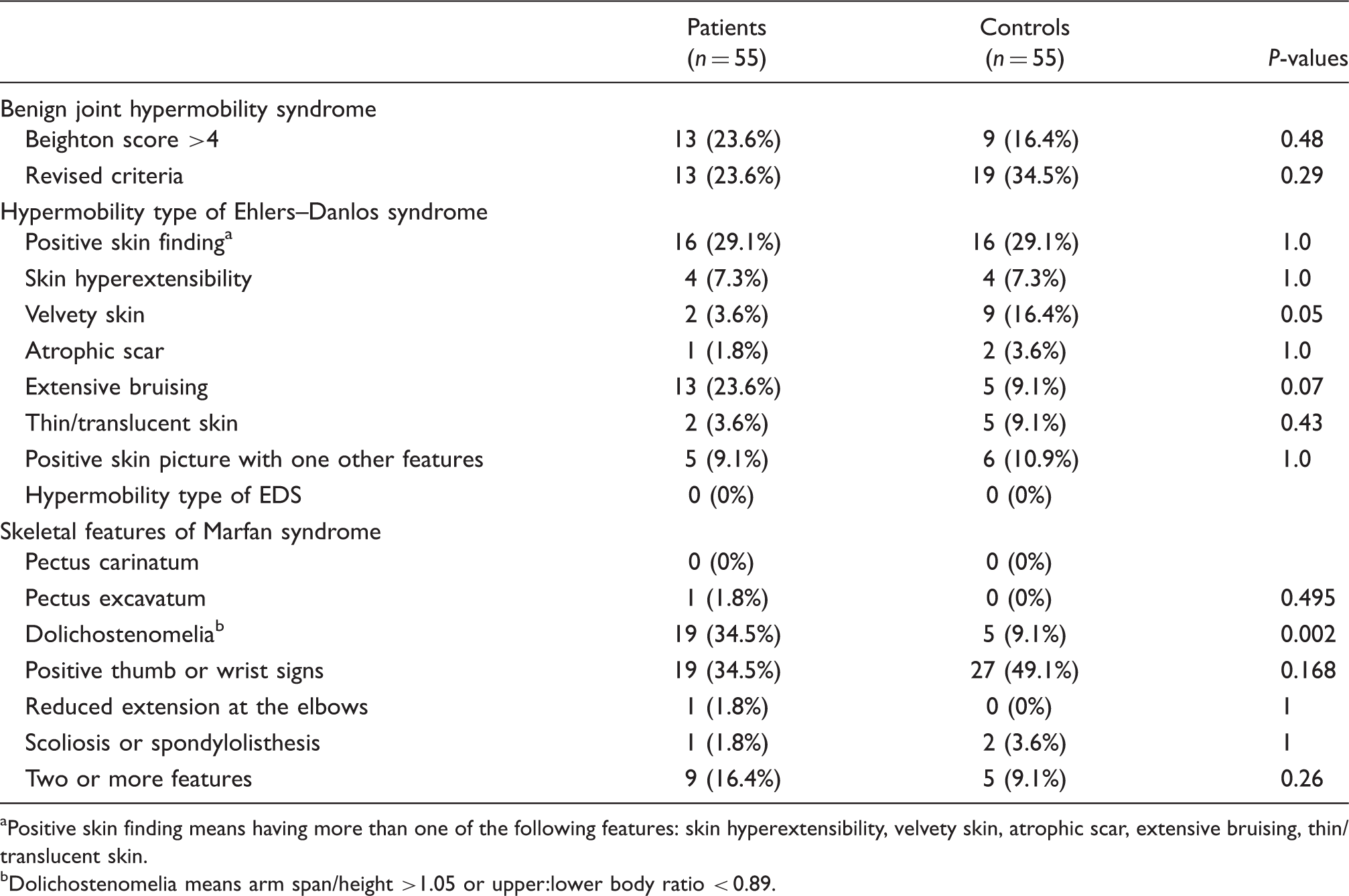
Positive skin finding means having more than one of the following features: skin hyperextensibility, velvety skin, atrophic scar, extensive bruising, thin/translucent skin.
Dolichostenomelia means arm span/height >1.05 or upper:lower body ratio < 0.89.
Extensive bruising was more commonly reported in patients than in the controls with a trend of statistical significance (SIH patients n = 13 [23.6%] vs controls n = 5 [9.1%]; P = 0.07). We found no difference between groups in the frequencies of other EDS skin features, including skin hyperextensibility, velvety skin, atrophic scarring, or thin and translucent skin. The frequencies of one or more EDS skin features were 29% (n = 16) in both patient and control groups. Five of the 16 SIH patients and six of the 16 controls (P = 1.0) also exhibited other features of hypermobility EDS, such as generalized joint hypermobility (Beighton score >5/9), recurrent joint subluxation and dislocation, chronic joint or limb pain, or family history. However, no case in either group fit the criteria for a diagnosis of the hypermobility type of EDS.
Nine of the SIH patients (16%) and five of the control subjects (9%) had positive skeletal features of Marfan syndrome (P = 0.26). The SIH patients had a significantly higher percentage of dolichostenomelia (disproportionately long limbs) than the control subjects (34.5% vs 9.1%; P = 0.002), but an insignificantly lower frequency of positive thumb or wrist signs (34.5% vs 49%; P = 0.168). Only one SIH patient had pectus excavatum, and none of the control group exhibited pectus carinatum or pectus excavatum. None of the patients or controls had a history of ocular or cardiovascular involvement with Marfan syndrome.
Discussion
A US study used Beighton hypermobility scores to determine that 11% of SIH patients demonstrated joint hypermobility, a prevalence that exceeded that of the general US population (5%) (3). Our SIH patients seemed to have a higher frequency (23.6%) of joint hypermobility than the control subjects (16.4%), but this difference was not significant. Of note, our control group had a similar prevalence of hypermobility as that found in a previous study (5) of Asian populations (17%). In contrast, the revised criteria for benign joint hypermobility syndrome indicate that the frequency of joint hypermobility in our SIH patients was 23.6%, but that for our controls was 34.5%. Therefore, the frequencies of joint hypermobility were higher in both our patient and control groups than in Western populations (6), which may be attributed to racial differences. However, our study did not confirm the finding that SIH patients had a higher frequency of joint hypermobility than that of a control group.
EDS is one of the most prevalent hereditary connective tissue disorders (1:5000 to 1:10,000) (10). The three main clinical manifestations of this disorder are skin hyperextensibility, joint hypermobility, and tissue fragility. More than 90% of EDS patients are classified as having type III hypermobility (10). One US study reported that 11% of SIH patients had classical type II EDS, with joint hypermobility, velvety skin, skin hyperextensibility, pes planus, and abnormal scarring (3). In our study, the frequencies of EDS skin features (29%) and other features of the hypermobility type of EDS (9%) in our SIH patients were almost the same as those in the control group (29% and 11%, respectively). Of note, none of our SIH patients fulfilled the criteria for a definitive diagnosis of a hypermobility type of EDS.
SIH has been associated with Marfan syndrome and meningeal abnormalities (12). Minor skeletal features of Marfan syndrome have been reported in 17% of SIH patients (3). However, we found no differences in the frequencies of positive Marfan skeletal features between the SIH patients and controls. The SIH patients exhibited only a higher frequency of dolichostenomelia than the control group (34.5% vs 9.1%; P = 0.002). Dolichostenomelia is not specific to Marfan syndrome, and this feature was not present in SIH patients in the study of Schievink et al. (13) One study of Asian populations showed that the mean arm-span-to-height ratio was higher in Asian males than in Caucasian males (1.03 vs 1.01; P < 0.01) (14). Therefore, ethnic differences may account for the presence of dolichostenomelia among our patients. Previous studies have also failed to demonstrate an association between the gene mutations of Marfan syndrome (FBN1 and TGFBR2) and SIH (13,15). Like the present study, these genetic studies have down-played the role of Marfan syndrome in SIH patients. Nevertheless, further studies are needed to elucidate the clinical meanings of a higher proportion of dolichostenomelia in patients with SIH when compared with controls.
Study limitations
Our study was limited by several factors. First, the exact frequencies of protrusio acetabuli and medial malleolus displacement, two skeletal anomalies of Marfan syndrome, could not be determined without radiography. In addition, the echocardiograms, slit lamp eye examinations, and genetic studies were not included in this study, which might have produced false-negative results of Marfan syndrome. Second, the control subjects were selected mainly from the hospital staff, which may have introduced some selection bias. Nevertheless, none of the control subjects had an individual or family history of SIH and the frequencies of connective tissue disorders in our control group were similar to previous studies conducted with Asian populations (5). Therefore, we concluded that the possibility of inflated frequencies of connective tissue disorders due to selection bias was very low. Third, the examiner was not blinded to the subject’s clinical status (controls vs SIH patients), which may have introduced observer bias. Fourth, even though we collected a large number of patients with SIH (n = 55) which is an uncommon headache, larger sample sizes are needed to show the statistical difference between SIH and control groups. For example, if the alpha error is 5% and statistical power is 0.8, we need 381 subjects in each group for extensive bruising and 275 for positive skeletal features of Marfan syndrome. These large sample sizes are not easily achieved for a single-centre study.
Conclusions
Except for dolichostenomelia, our study failed to confirm the association between the other connective tissue disorders and SIH. Further studies recruiting a larger sample size and in different ethnic groups are still warranted to delineate the role of connective tissue disorders for this headache disorder.
Footnotes
Acknowledgements
This study was supported, in part, by a grant from the Taipei Veterans General Hospital (V99C1-063) and the Ministry of Education (Aim for the Top University Plan), Taiwan.