
Abstract
The present study was conducted to determine the effect of acute (1 h) and chronic (daily dose for 30 days) paracetamol administration on the development of cortical spreading depression (CSD), CSD-evoked cortical hyperaemia and CSD-induced Fos expression in cerebral cortex and trigeminal nucleus caudalis (TNC). Paracetamol (200 mg/kg body weight, intraperitonealy) was administered to Wistar rats. CSD was elicited by topical application of solid KCl. Electrocorticogram and cortical blood flow were recorded. Results revealed that acute paracetamol administration substantially decreased the number of Fos-immunoreactive cells in the parietal cortex and TNC without causing change in CSD frequency. On the other hand, chronic paracetamol administration led to an increase in CSD frequency as well as CSD-evoked Fos expression in parietal cortex and TNC, indicating an increase in cortical excitability and facilitation of trigeminal nociception. Alteration of cortical excitability which leads to an increased susceptibility of CSD development can be a possible mechanism underlying medication-overuse headache.
Keywords
Introduction
Cortical spreading depression (CSD) is an electrical phenomenon of cerebral cortex which underlies the development of migraine aura. Accumulating evidence indicates the relationship between CSD development and vulnerability to headache attacks. For instance, Ayata and colleagues (1) showed that various migraine preventive agents, regardless of their formulae and therapeutic mechanisms, share the similar effect in reducing the frequency of CSD in experimental animals. They also demonstrated the relationship between degree of CSD suppression and dose and duration of treatment. On the other hand, the increase in cortical susceptibility to develop CSD was reported in serotonin (5-HT)-depleted animals (2). This finding may explain an increase in headache frequency in chronic daily headache, the condition in which 5-HT level is low (3). The reduction in threshold for the induction of CSD was reported in female mice (4). The gender difference in cortical excitability may contribute to an increased prevalence of migraine in women.
Analgesic over-consumption is known to worsen headache in patients with primary headaches – either migraine or tension-type headache. A survey in a tertiary headache centre in the US showed that overused substances included butalbital containing combination products, paracetamol, opioids, aspirin, ergotamine tartrate, and various triptans (5). The mechanism underlying this phenomenon is not well defined. Possible explanations include the sensitisation of central trigeminal nociceptive neurones, derangement of endogenous pain control pathway, etc. Based on the relationship between CSD development and clinical headache described above, it is possible that chronic analgesic exposure may cause headache deterioration by increasing the cortical excitability which leads to an increased susceptibility of CSD development.
The objectives of this study were to determine the effect of acute and chronic analgesic exposure on the development of CSD and CSD-evoked trigeminal nociception. Paracetamol was chosen for the study because this drug is widely used among patients with chronic headaches. In addition, since the change in vascular compartment is also important in the process of headache pathogenesis, the effect of analgesic administration on the CSD-evoked cortical hyperaemia was also investigated.
Materials and methods
Animals and drug treatment
Adult, male, Wistar rats weighing 200–250 g (National Animal Centre, Mahidol University, Thailand) were housed and maintained on normal rat food and tap water ad libitum under controlled environmental conditions. The study protocol was approved by the Ethical Committee of Faculty of Medicine, Chulalongkorn University (003/2551).
The study had two components. The first part was to determine the acute effect of paracetamol on CSD development. In this experiment, rats were divided into paracetamol-treated and control groups (10 rats each). A single dose of paracetamol (200 mg/kg body weight, intraperitoneally) was given to the treatment group whereas vehicle (12.5% of 1,2-propanediol in 0.9% sterile saline) of the same volume was given to the control. CSD was elicited at 1 h after paracetamol injection. The second experiment aimed at investigating the effect of prolonged paracetamol exposure on CSD development and CSD-evoked trigeminal nociception. Rats were divided into paracetamol-treated and control groups (10 rats each). Paracetamol (200 mg/kg body weight, intraperitoneally) or vehicle was administered once daily for the period of 30 days. Twenty-four hours after the last injection, animals were prepared for CSD induction.
In both experiments, CSD was induced in the parietal cortex by the topical KCl application method. Electrocorticograms were recorded continuously for 1 h using a glass micro-electrode. Cortical blood flow was monitored in frontal area using laser Doppler flowmetry. Two hours after CSD induction, rats were humanely killed. Brain and upper cervical cord were removed for Fos immunohistochemical study. The liver was also removed for histopathological examination using the standard haematoxylin and eosin method to exclude the hepatotoxicity induced by paracetamol. The presence of centrilobular or panacinar necrosis and sinusoidal congestion were used to indicate the paracetamol-induced hepatotoxicity.
Induction of CSD
Rats were anaesthetised with sodium pentobarbital (60 mg/kg body weight, intraperitoneally). Additional doses were given as required to maintain surgical anaesthesia based on testing of corneal reflex and response to tail pinch. Ventilation was assisted by a positive-pressure ventilator (rodent ventilator model 683, Harvard Apparatus, Holloston, Massachusetts, USA) via a tracheotomy opening. Arterial blood pressure was continuously recorded using a pressure transducer (Nihon model TP-300T, Nihon Khoden, Tokyo, Japan) inserted into the femoral artery. Rats were firmly positioned in a stereotaxic frame. A midline surgical incision was done and skin and soft tissue overlying the skull were removed. A 2-mm craniotomy was performed with the saline-cooled drill on the parietal bone (7 mm posterior and 1 mm lateral to bregma). Dura mater was carefully opened by microneedle to expose the cortical surface. Artificial cerebrospinal fluid (NaCl 118 mM, KCl 4 mM, Na2HPO4.H2O 1 mM, NaHCO3 25 mM, CaCl2.2H2O 1.5 mM, MgSO4.7H2O 1.2 mM, dextrose 5 mM in distilled water, pH 7.4, 37°C) was superfused to prevent the cortical surface from dryness. After all operation procedures had been completed, a single 3-mg KCl crystal was placed directly on the surface of parietal cortex to generate the CSD and was left on the cortical surface for the entire experimental period. This technique can generate a series of unifocal CSD. To avoid the variation in the CSD induction method, the amount of KCl, size of craniotomy well and amount of artificial cerebrospinal fluid were strictly controlled.
Electrocorticographic recording
To record cortical activity, another craniotomy (diameter 5 mm) was done at 1 mm anterior and 1 mm lateral to bregma. Microelectrode (internal diameter 5 µm) was prepared from bromosilicate glass, pulled with micropipette puller (Industrial Science Associates, Inc., Forest, Ridgewood, New York, USA). The micro-electrode was filled with NaCl (4 M) and then an Ag/AgCl wire was inserted. The completely filled glass micro-electrode was inserted perpendicular to the cortex to the depth of 500 µm from the cortical surface using an hydraulic micromanipulator (Narishige, Scientific Instrument Lab., Tokyo, Japan). An Ag/AgCl reference electrode was placed on the skin at the back. The obtained electrical signal was amplified using a micro-electrode amplifier (P16B; Grass Instrument, Quincy, Massachusetts, USA). The frequency band of electrophysiological acquisition was between 1–10 kHz. Due to the very high signal-to-noise ratio, the data filtration process was not necessary. Analogue data were converted to digital form using data acquisition system (PowerLab 4SP; Bella Vista, NSW, Australia). All tracings were analysed using PowerLab computer software (PowerLab 4SP). Measured variables included amplitude and area under the curve (AUC) of each CSD wave as well as the number of CSD waves occurring within a 1-h period.
Cortical blood flow recording
In order to monitor the CSD-evoked change in the cortical blood flow, the fibre-optic needle probe of a laser Doppler flowmeter (ALF 21; Advance Co. Ltd, Tokyo, Japan) was placed adjacent to the glass micro-electrode. The probe was inserted perpendicularly at a distance of 2 mm above the cortical surface. The wavelength of the laser beam was 780 nm. Data were recorded on a polygraph (Nihon RM 6000; Nihon Khoden, Tokyo, Japan). Peak amplitude of each hyperaemic cycle was calculated as percentage change from baseline value.
Fos immunohistochemical study
After completion of cortical activity and blood flow recording, rats were humanely killed by excessive dose of sodium pentobarbital and perfused transcardially with 250 ml of phosphate buffer, followed by 250 ml of 4% paraformaldehyde in 0.1 M phosphate-buffered saline (PBS), pH 7.4. Brain and cervical spinal cord were removed and immediately immersed in 4% paraformaldehyde in 0.1 M phosphate buffer. After overnight fixation, tissue was placed in a cryoprotectant solution (30% sucrose in 0.1 M phosphate buffer, pH 7.4) before being cut with a cryomicrotome at –20°C. Somatosensory cortex was identified according to the co-ordinates described by Paxinos and Watson. [6] and was coronally sectioned (30 µm thick). The caudal medulla (3 mm. caudal to the obex) to the first cervical cord was transversely cut and one in every five sections was collected. Sections were kept in cold 0.01 M PBS. All sections were incubated with 3% hydrogen peroxide in 50% ethanol for 30 min to minimise endogenous peroxidation. After repeated rinses in PBS, the sections were pre-incubated in PBS containing 3% normal horse serum, 1% bovine serum albumin for 60 min at room temperature and incubated overnight with rabbit anti-Fos (Santa Cruz Biotechology; (1:500 dilutions)). They were then incubated for 30 min with biotinylated goat anti-rabbit antiserum (Dako LSAB 2 system, Glostrup, Denmark) and rinsed again in PBS before incubating for 30 min in a streptavidin-horseradish peroxidase solution (Dako LSAB 2 system, Glostrup, Denmark). Bound peroxidase was revealed by incubating all sections in a solution containing 0.05% 3,3-diaminobenzidine, 0.005% hydrogen peroxide for 10 min. The reaction was stopped by repeated rinses in PBS. Sections were mounted on gelatinised slides, dehydrated in a graded series of ethanol, mounted, cover-slipped with Permount, and examined with a light microscope.
The number of Fos-immunoreactive (Fos-ir) cells was determined using image analysis software (ImagePro® Plus; Media Cybernetics Inc., Bethesda, Maryland, USA). To determine the number of Fos-ir cells in somatosensory cortex, a 250 × 250 µm square was drawn in layer V of the parasaggital part of this cortex and Fos-ir cells confined in the square were counted. Data from 10 areas sampled from each section (10 sections per rat) were averaged and expressed as number per 6.25 × 104 µm2. Expression of Fos in TNC was quantitated by counting Fos-ir neurones in lamina I and II of TNC from 10 sections of the cervical spinal cord and 10 sections from caudal medulla. The data were averaged and expressed as number per section. The identity of each rat was concealed throughout the counting process.
Statistical analysis
All cortical activity variables as well as numbers of Fos-ir neurones are expressed as mean ± SD. Possible statistically significant differences between paracetamol-treated and control groups was determined by Mann–Whitney U-test. Probability values of less than 0.05 were considered to be statistically significant.
Results
Exposure to paracetamol, acute and chronic, did not alter animal behaviour, including feeding as shown by the comparable body weights between the two groups. The averaged body weight of the chronic paracetamol-treated and control groups were 410 ± 20 g and 392 ± 29 g, respectively. The histological examination of liver was normal. There was no change in hepatocyte morphology or evidence of inflammation was observed.
Effect of acute paracetamol exposure
Effect on cerebral cortex
Cortical KCl application elicited a series of cortical depolarisations characterizing the CSD. Comparison between the two groups, showed the number of CSD waves generated in the first hour was similar (13.1 ± 1.1 and 13.7 ± 1.5 peak for control and paracetamol treated groups, respectively; P = 0.332). Despite the similarity in the frequency of CSD occurrence, the feature of individual DC shift observed in the control and paracetamol groups was different. The amplitude of DC shift was significantly greater in the paracetamol-treated group (31.5 ± 2.3 mV) as compared to the control (28.1 ± 3.3 mV; P = 0.001). Minimal change in CSD amplitude over time was observed with larger amplitude in the early waves. However, the difference was not statistically significant. The AUC of the DC shift was also greater in the paracetamol-treated group. The AUCs of the DC shift were 93.8 ± 24.7 and 73.4 ± 18.5 mV-s, for the paracetamol-treated and control groups, respectively; P = 0.006). The magnitude of increase was 12.8% from the baseline value (Figure 1 and Table 1). No significant difference was observed when the durations of DC shift between acute paracetamol and control were compared.
Effect of acute and chronic paracetamol administration on development of CSD (upper trace) and CSD-evoked cortical hyperaemia (lower trace). Acute paracetamol exposure leads to an increase in CSD amplitude and area-under-the curve without change in frequency (upper two panels). On the other hand, chronic exposure results in an increase in CSD frequency (lower two panels). A decrease in CSD-evoked cortical hyperaemia is observed in both acute and chronic paracetamol groups compared with the respective controls. The effect of acute and paracetamol administration on the development of CSD and CSD-evoked Fos expression in the cerebral cortex and TNC All variables were expressed as mean ± SD.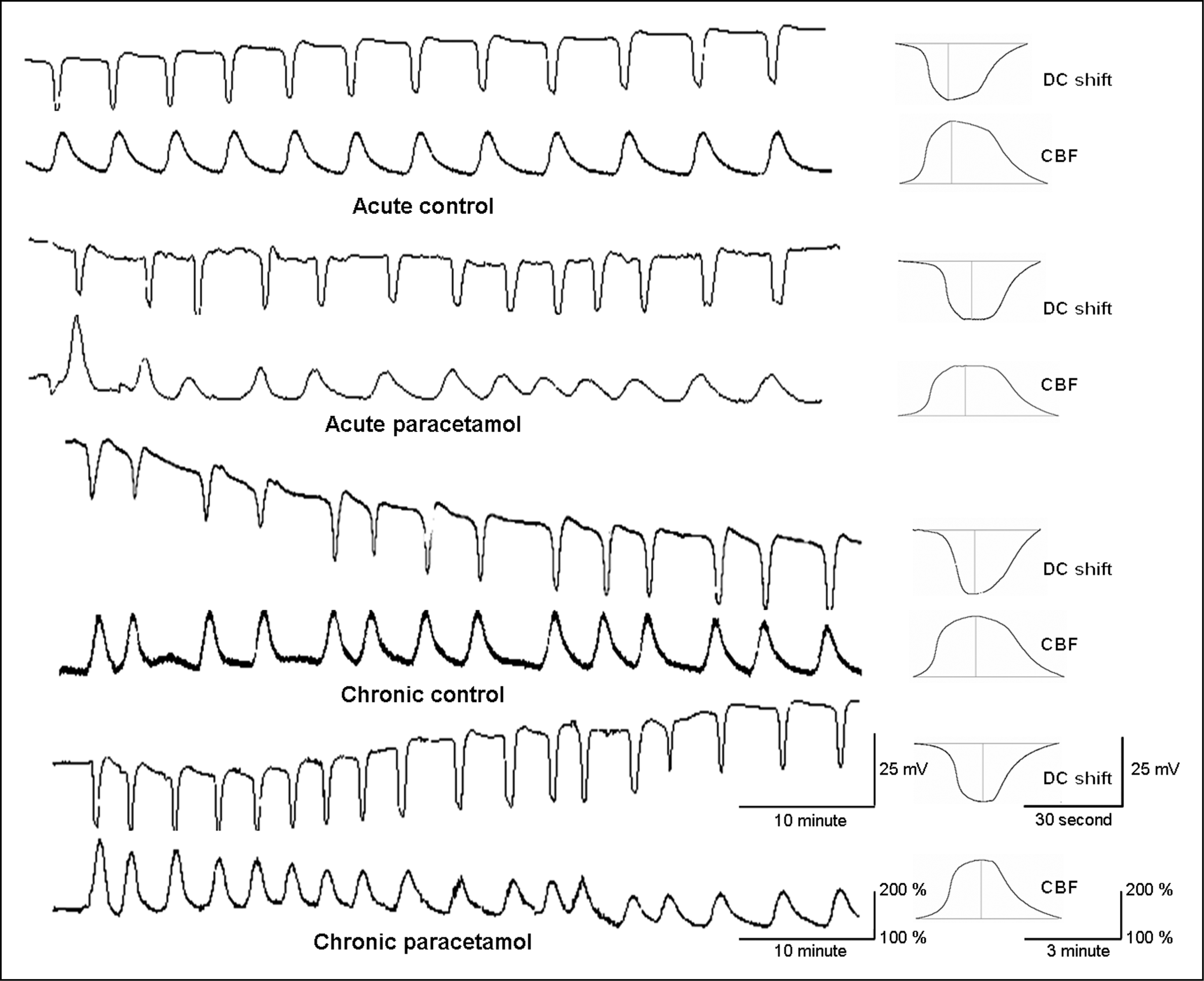

The cortical blood flow study revealed the cyclical hyperaemia which perfectly corresponded to the cycle of CSD. The rise in cortical blood flow usually occurred prior to the DC shift. Pretreatment with paracetamol lowered the degree of hyperaemia induced by CSD. The peak amplitude of CSD-evoked cortical hyperaemia observed in the paracetamol-treated and control groups were 185 ± 37% and 222 ± 62% from the baseline value, respectively. The difference between paracetamol-treated and control groups was statistically significant (P = 0.001).
The anti-Fos immunohistochemical study of cerebral cortex showed high density of Fos-ir neurones in the hemisphere ipsilateral to side of CSD induction. Only a few Fos-ir neurones were observed in the non-stimulated contralateral hemisphere. Pretreatment with paracetamol can reduce the density of CSD-evoked Fos expression in cerebral cortex. This reduction was more prominent in layer V. The density of Fos-ir cells in layer V in the paracetamol and control groups were 54.2 ± 16.1 and 113.9 ± 15.2 cells per 6.25 × 104 µm2, respectively (P = 0.026; Figure 2 and Table 1). The Fos-ir cells were also visualised in amygdala, dorsal raphe, thalamus, and hypothalamus (especially paraventricular nucleus). There was no significant difference between control and paracetamol groups.
Effect of acute and chronic paracetamol administration on CSD-evoked Fos expression in cerebral cortex. Acute treatment with paracetamol decreases the number of Fos-immunoreactive cells in layer 3 while chronic exposure increases CSD-evoked Fos expression. (A) Acute control; (B) acute paracetamol; (C) chronic control; (D) chronic paracetamol. Bar = 250 µm.
Effect on CSD-evoked trigeminal nociception
Application of KCl increased Fos expression in laminar I and II of the TNC especially in the ipsilateral side. The average numbers of Fos-ir cells in ipsilateral and contralateral TNC were 10.5 ± 2.2 and 4.9 ± 2.1 cells per slide, respectively. Pretreatment with paracetamol reduced the number of CSD-evoked Fos-ir neurones in both sides of TNC. The numbers of Fos-ir cells in the ipsilateral and contralateral TNC in the acute paracetamol treated group were 5.0 ± 2.3 and 2.4 ± 1.1 cells per slide, respectively. The difference between the paracetamol and control groups was statistically significant in both sides (P = 0.001; Figure 3).
Effect of acute and chronic paracetamol administration on CSD-evoked Fos expression in trigeminal nucleus caudalis. Acute treatment with paracetamol minimises the number of Fos-immunoreactive cells in the TNC, reflecting the anti-nociceptive effect. Conversely, chronic treatment enhances the CSD-evoked nociception as shown by an increase in the number of Fos-immunoreactive cells in the TNC. (A) Acute control; (B) acute paracetamol; (C) chronic control; (D) chronic paracetamol. Bar = 250 µm.
Effect of chronic paracetamol exposure
Effect on CSD and Fos-immunoreactivity in cerebral cortex
Chronic exposure to paracetamol increased the frequency of CSD waves. The number of CSD waves observed in 1 h from chronic paracetamol treated group and the control group were 15.8 ± 1.5 and 12.5 ± +1.3 peak per hour, respectively (P = 0.032). Unlike those observed in the acute treatment group, neither change in peak amplitude nor AUC was evident between chronic paracetamol-treated and control groups. The average amplitude and AUC of CSD waves from the chronic paracetamol group were 29.76 ± 3.38 mV and 65.38 ± 16.77 mV-s, respectively, while those of the control group were 27.30 ± 2.98 mV and 70.01 ± 19.84 mV-s, respectively (P = 0.100 and P = 0.355 for peak amplitude and AUC, respectively). The increase in CSD frequency was parallel to the increase in the density of Fos-ir cells in cerebral cortex. The number of Fos-ir cells in the chronic paracetamol and control groups were 113.9 ± 15.2 and 79.0 ± 17.7 per 6.25 × 104 µm2, respectively (P = 0.001; Figure 1 and Table 1).
Similar to results observed in the acute paracetamol experiment, the cortical blood flow study showed significant lower degree of CSD-induced hyperaemia in the animals receiving chronic paracetamol administration. The peak amplitude of CSD-evoked hyperaemia in the chronic paracetamol and control groups were 199 ± 49% and 244 ± 43% from baseline value, respectively (P = 0.010).
Effect on CSD-evoked trigeminal nociception
In the chronic paracetamol treated group, the average number of Fos-ir cells in the ipsilateral and contralateral sides were 16.4 ± 2.9 and 10.2 ± 3.2 cells per slide, respectively. The numbers of Fos-ir cells in the ipsilateral and contralateral TNC in the control group were 11.5 ± 2.8 and 7.4 ± 3.1 cells per slide, respectively. The number of Fos-ir cells in the chronic paracetamol treated group was significantly higher than control group (P-values 0.04 and 0.015 for ipsilateral and contralateral sides, respectively; Figure 3).
Discussion
The present study shows that the trigeminal system responds differently to acute and chronic analgesic exposure. Short-term exposure to paracetamol reduces the number of CSD-evoked Fos-ir neurons in the TNC without alteration in CSD frequency. On the other hand, chronic exposure leads to an increase in CSD frequency and CSD-evoked Fos expression in the cerebral cortex and the TNC. Concerning cortical blood flow, paracetamol reduces the amplitude of CSD-evoked cortical hyperaemia regardless of the duration of exposure.
CSD is believed to be the physiological mechanism underlying the aura phase of migraine. Changes in CSD frequency may reflect the deterioration in the excitability of cerebral cortex and susceptibility to develop migraine attacks. Several factors have been reported to affect the frequency of CSD waves. In general, factors or conditions that reduce neuronal excitability will reduce the frequency of CSD while those which increase the excitability will increase the CSD frequency. For instance, CSD frequency was reduced in rats treated with anaesthetics such as halothane, isofurane and sevoflurane (7). N2O can reduce CSD frequency when combined with isofurane or urethane, but not α-chloralose(8). Reduction in CSD frequency has also been observed in animals chronically treated with various classes of anti-migraine preventive medications (1). On the other hand, the high frequency of CSD as well as the increase in the trigeminal nociception has been reported in animal with low serotonin (2). Several lines of evidence indicate that the development of CSD can affect the synaptic transmission in cerebral cortex. Using an ex vivo brain slice technique, Wernsmann et al. (9) demonstrated that tetanus-induced long-term potentiation was significantly enhanced in hippocampal slices obtained from the ipsilateral site to the hemisphere in which CSD was evoked. A more recent study with a human brain slice model also showed that the induction of CSD significantly increased the amplitude of field excitatory post-synaptic potential and increased the induction of LTP in the third layer of neocortical tissues (10). These findings are compatible with our observation of an increase in CSD-evoked Fos expression in cerebral cortex especially in those with greater numbers of CSD waves. All this evidence indicates the facilitating effect of CSD on synaptic excitability which may contribute to neocortical hyperexcitability. Extending this hypothesis, an increase in the number of CSD waves occurring in the prolonged paracetamol-treated group may further enhance the excitability of the cortex, resulting in the cortical hyperexcitability state.
Besides frequency, changes in other CSD variables have also been reported. The reduction in threshold and increases in propagation velocity of CSD have been demonstrated in R192Q knock-in mice, an animal model for familial hemiplegic migraine (11). The recent study by Tong and Chesler (12) has revealed that acidosis can increase the threshold for CSD induction but decrease the duration and velocity of CSD. Oestrogen and progesterone have been reported to enhance the repetition rate as well as the amplitude of spreading depression in neocortical slices treated with hypotonic artificial cerebrospinal fluid or KCl micro-injection (13). In this study, we observed an increase in the amplitude of CSD without frequency change in rats treated with a single dose of paracetamol. The increase in CSD amplitude disappeared in rats with chronic paracetamol administration. Although the mechanism by which acute paracetamol exposure increases the CSD amplitude remains unclear, alteration in astrocyte functions is a possible explanation. It is known that astrocytes play an important role in the regulation of extracellular calcium and potassium as well as transmitters, especially glutamate, during CSD. Therefore, derangement in astrocyte function can alter the cortical excitability. The mutation in the Na+/K+-ATPase pump gene ATP1A2 expressed in astrocytes has been reported to associate with familial hemiplegic migraine (14). Effects of paracetamol on some astrocyte functions have been reported. For instance, Mancini et al. (15) demonstrated that paracetamol can down-regulate the nuclear translocation of nuclear factor-kappa B and inhibits prostaglandin E2 production. However, information concerning the effect of paracetamol on extracellular ion regulation of astrocytes is still unavailable.
In this study, we observed the dissociation between amplitude of CSD and degree of cortical hyperaemia. In the acute group, the increase in CSD amplitude co-existed with the decreased degree of CSD-induced cortical hyperaemia. The decrease in CSD-evoked cortical hyperaemia was also evident in chronically treated animals without changes in CSD amplitude. The alteration in CSD-evoked cortical hyperaemia may be the result of a direct effect of paracetamol on cerebral circulation. The mechanism underlying the CSD-evoked cortical hyperaemia is complex and involves several chemical messengers (16). Some of these messengers can be affected by paracetamol. For instance, it has been shown that paracetamol can affect cerebral endothelial cells causing the inhibition of prostaglandin production (17). Anti-oxidant and anti-inflammatory effects of paracetamol on the cerebral vasculature have also been demonstrated [18). These effects may alter cerebrovascular motor tone and diminish the degree of cortical hyperaemia evoked by CSD.
The present findings lead to two further questions, what is the mechanism underlying these changes and whether these observations can be generalised to other analgesics. Recently, Mallet et al. (19) have suggested roles for the endocannabinoid and serotonin systems in the anti-nociceptive mechanism of paracetamol. They proposed that the analgesic effect of paracetamol is mediated via the action of its metabolite AM404 upon the CB(1) receptors. Activation of the endocannabinoid system will reinforce the serotonergic bulbospinal pathways and attenuate the nociceptive process in the spinal cord. Serotonin also plays a role in paracetamol-induced analgesia by increasing transcript and protein levels of low-affinity neurotrophin receptor, insulin-like growth factor-1 (IGF-1) receptor alpha subunit, and growth hormone receptor. This neurotransmitter can reduce the amount of somatostatin receptor (sst3R) mRNA via the action of spinal 5-HT1A receptors (20). The involvement of serotonin in paracetamol-induced analgesia is supported by the observation that pretreatment with serotonin receptor antagonists can attenuate the analgesia induced by paracetamol (21). This serotonin-dependent nociceptive inhibition is also evident in humans. In 2006, Pickering et al. (22) showed that co-administration of paracetamol with tropisetron or granisetron, 5-HT3 antagonists, completely blocked the analgesic effect of paracetamol in humans. It has been proposed that paracetamol may act centrally by reinforcing the descending inhibitory pain pathway (23). Our previous experiments also support the role of serotonin in the analgesic mechanism of this agent. Acute exposure to paracetamol increases 5-HT levels and down-regulates the pro-nociceptive 5-HT2A receptor whilst chronic exposure decreases the 5-HT level and up-regulates this receptor (24). The reverse relationship between 5-HT level and its receptors reflects the fact that receptor up-regulation may be secondary to the reduction in the transmitter level. Since activation of 5-HT2A receptor leads to an increase in neuronal excitability, an up-regulation of this receptor in cerebral cortex may increase the cortical neuronal response. This mechanism may explain the increased CSD response and cortical Fos expression observed in rats following chronic treatment with paracetamol in this study. Of note, the alteration in 5-HT2A receptor expression cannot explain the effect of paracetamol in attenuating the CSD-evoked cerebral hyperaemia. In addition to neural tissue, 5-HT2A receptors are also expressed in cerebral vessels (25). This vascular receptor is likely to mediate the vasodilator effect (26). Therefore, up-regulation of this receptor would increase cerebral blood flow rather than decrease it, as shown in this study. To our knowledge, there is no study investigating the effect of paracetamol on vascular 5-HT2A receptor expression.
Concerning the issue of generalisability, clinical observations show that medication-overuse headache can result from the prolonged use of analgesics, ergots or triptans. The medication-overuse headaches share common clinical features regardless of the variety of its causative agents. This observation suggests that all drugs may share a common mechanism. This hypothesis is supported by the observation that chronic exposure to triptans also alters the central serotonin-dependent pain controlling system (26). Therefore, it is possible that chronic use of other acute anti-migraine medications may increase cortical excitability as observed in rats with prolonged paracetamol exposure. It should be noted that, although paracetamol has been reported as a common drug used by chronic headache sufferers, it is mostly used in combination with other compounds such as opioid analgesics. Therefore, more studies with other classes of medication are needed to confirm this hypothesis.
Conclusions
The present study demonstrates the plasticity of the cerebral cortex and trigeminal nociceptive system in response to acute and chronic analgesic exposure. While acute exposure elicits the anti-nociceptive effect, chronic administration of analgesics increases the excitability of cerebral cortex as well as facilitating the trigeminal nociceptive mechanism. The temporal profile resembles the clinical features of chronic headache observed in patients with analgesic-induced headache. Therefore, neuronal hyperexcitability may explain the development of chronic headache in these patients.
Footnotes
Acknowledgements
This study was supported by the 90th Anniversary of Chulalongkorn University Fund (Ratchadphiseksomphot Endowment Fund), Chulalongkorn University and the Thailand Research Fund (RTA5180004).