
Abstract
Objective
The objective of this article is to evaluate electrically evoked thresholds for cortical spreading depression (CSD) and stress-induced activation of trigeminal afferents in a rat model of medication-overuse headache (MOH).
Methods
Sumatriptan or saline was delivered subcutaneously by osmotic minipump for six days to Sprague-Dawley rats. Two weeks after pump removal, animals were anesthetized and recording/stimulating electrodes implanted. The animals were pretreated with vehicle or topiramate followed by graded electrical stimulation within the visual cortex. CSD events were identified by decreased EEG amplitude and DC potential shift. Additional unanesthetized sumatriptan or saline-pretreated rats were exposed to bright light environmental stress and periorbital and hindpaw withdrawal thresholds were measured. Following CSD stimulation or environmental stress, immunohistochemical staining for Fos in the trigeminal nucleus caudalis (TNC) was performed.
Results
Sumatriptan pre-exposure significantly decreased electrical stimulation threshold to generate a CSD event. Topiramate normalized the decreased CSD threshold as well as stress-induced behavioral withdrawal thresholds in sumatriptan-treated rats compared to saline-treated animals. Moreover, CSD and environmental stress increased Fos expression in the TNC of sumatriptan-treated rats, and these effects were blocked by topiramate. Environmental stress did not elicit cutaneous allodynia or elevate TNC Fos expression in saline-treated rats.
Conclusions
A previous period of sumatriptan exposure produced long-lasting increased susceptibility to evoked CSD and environmental stress-induced activation of the TNC that was prevented by topiramate. Lowered CSD threshold, and enhanced consequences of CSD events (increased activation of TNC), may represent an underlying biological mechanism of MOH related to triptans.
Keywords
Introduction
Migraine is characterized by an intense, typically unilateral headache usually accompanied by heightened sensitivity to light, sound or movement along with nausea and vomiting (1). Growing evidence suggests that cortical spreading depression (CSD) may be the bioelectrical homologue of migraine aura (2–7). A CSD event generally manifests as depressed electrocortical activity propagating from a brain region at a rate of 3–6 mm/min, which coincides with the progression of visual scotoma seen in migraine with aura (8). CSD has also been shown to activate dural trigeminal afferents, providing a potential pathophysiological basis for the classic clinical observation of aura followed by migraine headache (9,10).
Medication-overuse headache (MOH) related to the frequent consumption of triptans and other acute medications is a well-recognized clinical condition that is frequently responsible for the progression of migraine from an episodic to chronic disorder with very frequent headache (11–14). Patients with migraine complicated by MOH demonstrate a higher prevalence of cutaneous allodynia (CA) than patients with episodic migraine (15–17). We previously showed that rats develop CA during sustained or intermittent triptan exposure (18,19). Additionally, after termination of triptan exposure and normalization of baseline sensory thresholds, triptan-pretreated rats remained sensitive to putative migraine triggers (e.g. nitric oxide (NO) donor or environmental stress), suggesting a state of “latent sensitization” (18). The reasons for enhanced sensitivity to putative migraine triggers are not understood but one possibility is that these triptan pre-exposed rats may have enhanced susceptibility to generation of CSD events with subsequent activation of trigeminal dural afferents. Such long-lasting changes induced by triptans would be consistent with the increased frequency of migraine headaches in patients with MOH. In the present study we measured electrically evoked CSD thresholds and potential activation of trigeminal afferents following CSD or environmental stress in a rat model of triptan-induced MOH. Additionally, we determined if these changes could be reversed by treatment with topiramate, as a proposed mechanism of action of topiramate is its ability, demonstrated after repeated injections, to raise the threshold for evoked CSD events (20).
Materials and methods
Animals
Male Sprague-Dawley rats (150–175 g, Harlan) were housed in a climate-controlled room with a 12-hour light/dark cycle (lights on at 7:00 a.m.) with food and water ad libitum. Studies were performed while animals were on their light cycle between the times of 7:00 a.m. and 7:00 p.m. All procedures were performed in compliance with the National Institutes of Health (NIH) guidelines for use of laboratory animals and with the regulations of the Institutional Animal Care and Use Committee (IACUC) of the University of Arizona. Behavioral studies were performed by an experimenter blinded to the treatment conditions.
Induction of latent sensitization
Rats were maintained under light anesthesia (2% isoflurane in room air at 1 l/minute) and surgically implanted (Day 0) with osmotic minipumps (Alzet, Cupertino, CA, USA; model 2001) to constantly deliver sumatriptan (0.6 mg/kg/day) or 0.9% saline at a nominal flow rate of 1 µl/hour for seven days (21). Sumatriptan was provided by Neuraxon Inc. The minipumps were removed on Day 6 under isoflurane anesthesia.
Recording of electrical stimulation-induced CSD events
On Day 20 (i.e. 14 days after minipump removal), rats were anesthetized with divided doses of urethane until surgical anesthesia was obtained (mean dose: 1.8 g/kg, intraperitoneally (i.p.)). Urethane was used to provide very long-lasting anesthesia without significant effect on CSD initiation and propagation (22). Anesthetized rats were mounted in a stereotaxic headholder and body temperature was maintained at 37℃ with a feedback-controlled heating pad. As described previously (23), the skull was exposed and burr holes made to place the 1 mm ball tips of Ag/AgCl recording electrodes on the dura of the frontal and parietal cortices (2.0 mm right lateral, 1.5 mm anterior and 2.5 mm posterior to bregma, respectively). The reference electrode was located on the midline, 11.5 mm posterior to bregma or over the left hemisphere (6.5 mm posterior and 3 mm lateral to bregma). The free ends of the electrodes were soldered to a multipin connector (Continental Connector, Hatfield, PA). A pair of coated tungsten electrodes, with tips 1 mm apart (A-M systems, WA, 98382) was inserted 1.2 mm into cortical tissue through a burr hole over the visual cortex. Two small machine screws were threaded into the skull and the entire electrode assembly was secured with acrylic. The animal was then placed on a feedback-controlled heating pad in a Faraday cage for electroencephalographic (EEG) recordings. The presence of EEG burst suppression was used as an indicator of anesthetic overdose; consequently, animals showing burst suppression were not used in the study.
Electrical signals were acquired through a DC amplifier (15A12, Grass Instruments) to process DC potentials and an AC amplifier (15A54, Grass Instruments) to process the EEG potentials (23). Data were digitized at 100 Hz and recorded with Gamma v.4.9 (Astro-Med Inc) and analyzed offline. Recordings were begun at least 30 minutes prior to any manipulations to allow stabilization and to provide for a refractory period for any CSDs that might have been initiated as a result of surgery (20). Thirty minutes prior to initiating the electrical stimulation protocol, rats received an i.p. injection of either vehicle or topiramate (Janssen Pharmaceuticals Inc, Raritan, NJ), which was obtained from the pharmacy and prepared as a suspension (24). Electrical stimulation (Isolated Pulse Stimulator Model 2100; A-M Systems) was applied at four-minute intervals with charge increasing from 10 µC to 7000 µC by varying both current intensity and duration (24). The stimulation began with 1 mA for 10 msec and increased in steps of 25, 50, 100, 200, 300, 400 msec, then at 2 mA for 300, 800, 1000 msec, 3 mA for 400 msec, 4 mA for 400, 500, 1000 msec, 5 mA for 1000 msec, 6 mA for 1000 msec, and 7 mA for 1000 msec (24). The stimulation protocol was terminated once a CSD occurred. CSD events were identified by a marked reduction in EEG amplitude along with a corresponding change in membrane potential (i.e. DC shift) (25). If a CSD did not occur then the animal was assigned a threshold corresponding to the highest charge obtained.
Behavioral testing for tactile sensitivity
Behavioral thresholds to evoked stimuli were determined by applying calibrated von Frey filaments perpendicularly to the periorbital region and the plantar aspect of the hindpaw until a withdrawal response was elicited. The 50% withdrawal thresholds were determined by the Dixon “up and down” method (21,26–28). Additional groups of rats were exposed to bright light for one hour while in their home cages. A pair of halogen shop lights were placed 350 cm from both of the larger sides of the clear Plexiglas cage to deliver between 1300 lux to 1500 lux, approximating full daylight. With these parameters, temperature change inside the cage varied by less than 1℃. Periorbital and hindpaw sensory thresholds were measured every hour for six hours after exposure. The bright light stress was repeated 24 hours after the first exposure and behavioral sensory thresholds were assessed at hourly intervals for 6 hours.
Immunohistochemistry for trigeminal nucleus caudalis (TNC) Fos
Two hours after eliciting either a CSD or after bright light stress, the anesthetized rats were perfused, transcardially, with 1X phosphate-buffered saline and then 4% paraformaldehyde. Coronal sections (30 µm thick) of the caudal medulla were permeabilized with 0.2% TritonX100, blocked with 5% normal goat serum and incubated overnight with rabbit polyclonal anti-Fos (Santa Cruz, SC-52, 1:25,000). The sections were incubated with biotinylated anti-rabbit antibody followed by the ABC complex (Vectastain Elite ABC kit, Vector Labs) and Tyramide Signal Amplification detection (TSA Plus Fluorescein Kit, Perkin Elmer). Slides were mounted in Vectashield mounting medium (Vector Lab Inc). Photomicrographs of 10 to 15 caudal medullary sections per rat, at increments of 100 µm, were examined with ImageJ, by an experimenter blinded to the treatments, for Fos-positive nuclear puncta within the outer laminae of the TNC ipsilateral to the cortical hemisphere receiving electrical stimulation. The numbers of Fos-positive profiles found per section was recorded, and data presented as median and interquartile range for each treatment group.
Statistical analyses
CSD stimulation thresholds were compared among groups with the Kruskal-Wallis test followed by Dunn's Multiple Comparison Test to identify significant differences among groups, as were the Fos counts in TNC sections. The behavioral paradigms, DC shifts and propagation speeds were analyzed by analysis of variance (ANOVA) followed by Tukey's correction for multiple comparisons. In addition, Fast Fourier Transform (FFT) analyses were performed on EEG traces taken at 60-second intervals prior to CSD and centered within the CSD in order to detect changes in EEG amplitude and power. Comparisons for the stimulation threshold to initiate a CSD, DC deflection, CSD duration, propagation time, spectral power, and amplitude among the treatment groups were analyzed by ANOVA followed by Tukey's correction for multiple analysis.
Results
Sumatriptan infusion produces latent sensitization
Confirmation of CA in response to sumatriptan infusion (0.6 mg/kg/day) was indicated by the significant (p < 0.05) reductions in hindpaw withdrawal thresholds on Day 6 (Figure 1). Removal of the osmotic minipumps delivering sumatriptan resulted in a normalization of behavioral responses to light tactile stimuli evident on Day 20 (Figure 1). Infusion of saline vehicle did not reduce withdrawal thresholds on Day 6 (Figure 1).
Rats received infusions of saline or sumatriptan for six days when the osmotic minipumps were removed. Paw withdrawal thresholds were determined prior to minipumps implantation (Day 0), on Day 6 and 2 weeks after minipumps removal (Day 20). Paw withdrawal thresholds of the sumatriptan-exposed rats were significantly (*; p < 0.05) reduced from the Day 0 baseline value on Day 6. The withdrawal thresholds were not different from baseline by Day 20. Paw withdrawal thresholds of the saline-treated rats did not change over time.
Electrical stimulation threshold to induce CSD
The median stimulation threshold to induce CSD events in anesthetized saline-pretreated, vehicle-challenged rats was 5500 µC (4000 µC to 6000 µC). Prior exposure to sumatriptan produced a significant (p = 0.001) reduction in median CSD threshold to 600 µC (600 µC to 3760 µC) (Figure 2). Pretreatment with topiramate (80 mg/kg, i.p.) 30 minutes prior to initiation of electrical stimulation produced a significant (p = 0.001) elevation in the median CSD threshold of the sumatriptan-infused groups to 5000 µC (4000 µC to 6000 µC) (Figure 2). Of the 26 rats used in this part of the study, CSD events were elicited by electrical stimulation in 67% of saline-pretreated, vehicle-challenged rats (N = 6), 100% of sumatriptan-exposed, vehicle-treated rats (N = 6), 58% of saline-exposed, topiramate-challenged rats (N = 7), and 86% of sumatriptan-exposed, topiramate-challenged rats (N = 7). These observations are consistent with increased resistance to excitation after topiramate exposure. The mean change in membrane potential coincident with parietal and frontal CSD events was not significantly different among the groups (p > 0.05). The parietal DC shifts ranged from 1.7 ± 0.21 mV to 2.4 ± 0.30 mV and the frontal DC shifts ranged from 2.0 ± 0.15 mV to 2.9 ± 0.18 mV. The propagation speeds of the CSD events were not significantly different among the treatment groups (p > 0.05) and ranged from 4.11 ± 0.38 mm/sec to 6.59 ± 1.57 mm/sec. Depression of cortical activity was confirmed by significant reductions in EEG amplitude and power during the CSD event. However, both the baseline and mid-CSD amplitude and power values of the CSD events at either the frontal or parietal sites did not differ significantly (p > 0.05) among the groups.
(a) Rats received infusions of saline or sumatriptan for six days and allowed to recover. On Day 20, the animals received either vehicle or topiramate (80 mg/kg, i.p.) and electrical stimulation was applied in increasing charge levels until CSD was generated. Sumatriptan-exposed rats show a significant (p < 0.001) decrease in CSD stimulation threshold. Topiramate, given one hour prior to electrical stimulation, produced a significant (p < 0.001) elevation in median stimulation threshold compared to the sumatriptan-vehicle group. (b) Sample recording trace of EEG and DC membrane potentials.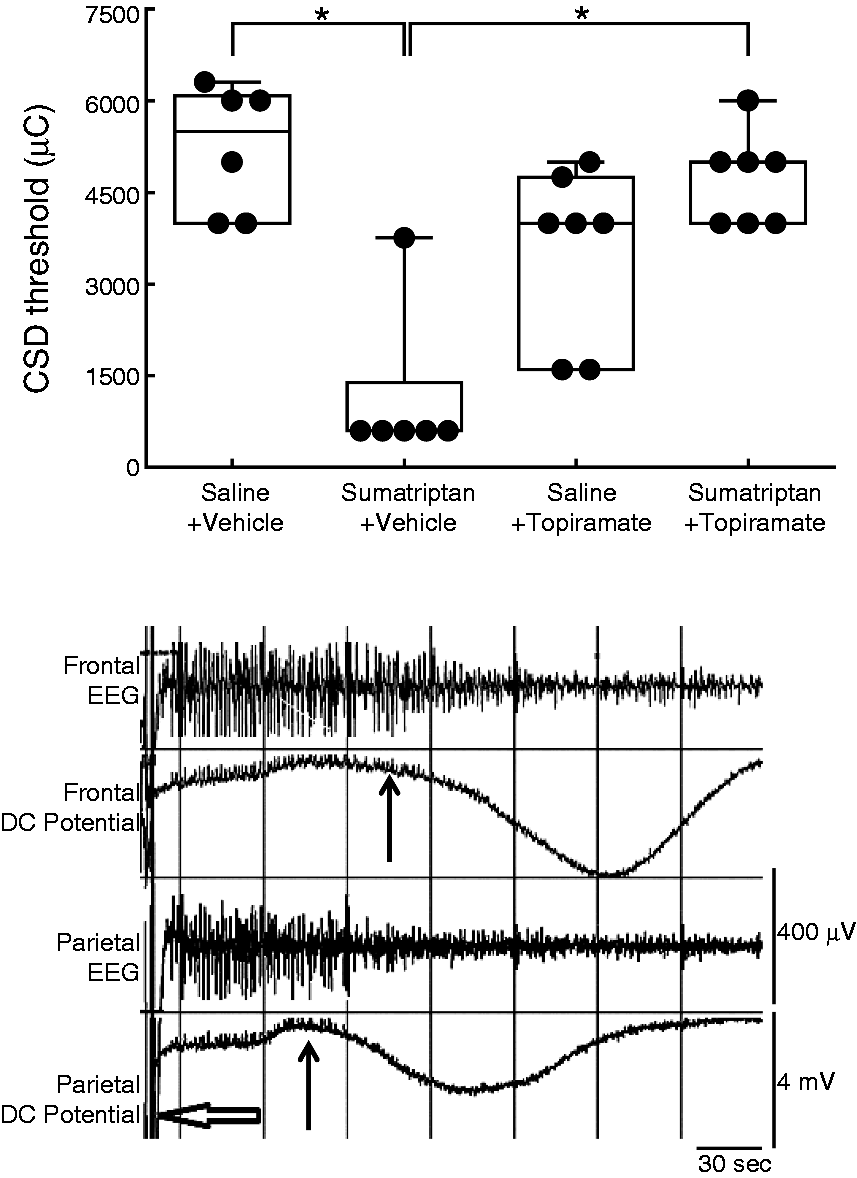
Sumatriptan exposure increases CSD-induced Fos expression in the TNC
Saline and sumatriptan-exposed rats that were anesthetized and prepared for CSD recording but without receiving electrical stimulation showed very little Fos expression in the TNC (Figure 3). Generation of a CSD event produced Fos expression both in saline- and sumatriptan-exposed rats that received vehicle injection 30 minutes prior to stimulation (Figure 3). However, TNC Fos was significantly greater in the sumatriptan-exposed rats compared to the saline-infused rats (p < 0.05). Topiramate (80 mg/kg, i.p.) given 30 minutes prior to CSD blocked the enhanced expression of TNC Fos in sumatriptan-exposed rats. The expression of Fos was not significantly different in saline- and sumatriptan-exposed rats that received topiramate (Figure 3).
Rats received infusions of saline or sumatriptan for six days and allowed to recover. On Day 20, the animals received either vehicle or topiramate (80 mg/kg, i.p.) and electrical stimulation was applied until CSD was generated. The animals were kept anesthetized for an additional two hours and were then perfused and prepared for immunohistochemical analyses. TNC sections were obtained and labeled for Fos. CSD produced an increase in median Fos expression in the TNC of rats that received saline or sumatriptan infusions compared to those receiving no stimulation (p < 0.05). The increase in Fos expression was significantly (p < 0.05) greater in the sumatriptan-infused rats. Pretreatment with topiramate blocked the elevated expression of Fos in the TNC. The median is indicated in each box and whisker plot within the box; the extents of the boxes represent the 25% and 75% quartiles, and the whiskers show the 10% and 90% limits. The dots represent data points falling outside these limits. The photomicrographs indicate a representative section of the TNC. The area indicated by the dotted lines indicates the region of interest for counting Fos profiles. Insets, indicated by the rectangle, show magnified views of the Fos label within the TNC. The original images captured by camera were color-inverted to better visualize the background of the sections and the Fos label.
Sumatriptan pre-exposure produces CA
A total of 30 rats were used in this part of the study. Saline-exposed rats receiving either vehicle (N = 6) or topiramate challenge (N = 9) 30 minutes prior to bright light environmental stress did not demonstrate CA (Figure 4). In contrast, sumatriptan-exposed animals that received vehicle (N = 6) and bright light stress showed significant (p < 0.05) reductions in periorbital and hindpaw withdrawal thresholds (Figure 4), suggesting the presence of latent sensitization (Figure 4). Topiramate (N = 9) blocked these behavioral signs of CA (Figure 4).
Saline and sumatriptan-treated rats received either vehicle or topiramate (80 mg/kg, i.p.) one hour prior to exposure to bright light environmental stress. Periorbital and hindpaw withdrawal thresholds were measured each hour for six hours. Sumatriptan-treated rats receiving vehicle demonstrated cutaneous allodynia indicated by a significant (p < 0.05) reduction in withdrawal thresholds over the observation period compared to the saline-/vehicle-treated group (two-factor ANOVA). Topiramate injection to the sumatriptan-treated rats resulted in a significant increase in withdrawal thresholds relative to the sumatriptan/vehicle group (two-factor ANOVA).
Exposure to bright light stress produced a significantly (p < 0.05) greater increase in Fos expression in the TNC of sumatriptan-exposed rats (N = 4) compared to the saline-exposed rats (N = 4) (Figure 5). Topiramate blocked the enhanced Fos expression observed in the TNC of sumatriptan-exposed animals (N = 5) (Figure 5). Moreover, topiramate reduced the stress-induced Fos expression in saline-exposed rats (N = 5), although this decrease was not significant (p > 0.05).
Rats exposed to saline or sumatriptan and that were subjected to bright light environmental stress were anesthetized two hours after the light exposure. The animals were perfused and the brains prepared for histochemical analyses of Fos expression. Sumatriptan exposure resulted in a significant (p < 0.05) increase in TNC Fos expression. This increase was abolished by pretreatment with topiramate (p < 0.05). The median is indicated in each box and whisker plot within the box; the extents of the boxes represent the 25% and 75% quartiles, and the whiskers show the 10% and 90% limits. The dots represent data points falling outside these limits. The photomicrographs indicate a representative section of the TNC. The area indicated by the dotted lines indicates the region of interest for counting Fos profiles. Insets, indicated by the rectangle, show magnified views of Fos label within the TNC. The original images captured by camera were color-inverted in order to better visualize the background of the sections and the Fos label.
Discussion
MOH is a common clinical disorder that results in increased frequency of headaches following the overuse of acute medications for an underlying primary headache disorder, most commonly migraine (12,14). Indeed, acute medication overuse is considered to be the most common underlying cause of chronic migraine, a highly disabling neurological disorder that affects approximately 2% of the population (12,14,29,30). Medications reported to increase the risk of MOH include butalbital and opioid-containing medications, mixed analgesics, ergotamine tartrate, nonsteroidal anti-inflammatory drugs (NSAIDs), and triptans (31,32). Whereas MOH does not exclusively manifest as migraine headache, the overuse of triptans produces either increased frequency of migraine or migraine-like headaches in a large majority of patients with triptan-related MOH (29,30,33). While the precise mechanisms that can lead to MOH are unknown, it is likely that the development of central sensitization plays a key role (11,34). Individuals with MOH have a greater prevalence of CA, supporting the likelihood that MOH represents a state of central sensitization (15,19,35). Central sensitization manifests clinically by the time-dependent development of CA that can spread to extracephalic regions during the course of a migraine episode (36–38). Imaging studies suggest that extracephalic CA reflects the sensitization of dural-specific trigeminovascular neurons in the sensory thalamus (39,40). Overuse of triptans or of NSAIDs increases cortical excitability in patients who develop MOH (41,42).
Our previous studies in rats demonstrated that 14 days following a period of triptan exposure, an increased number of cell bodies projecting to the dura mater were positively labeled for calcitonin gene-related peptide (CGRP) and neuronal nitric oxide synthase (nNOS) in spite of baseline sensory thresholds (18,21). Challenge of the animals with environmental stress or NO donor, widely considered to be “triggers” of migraine, resulted in a generalized (facial and hindpaw) CA that was delayed, peaking two to three hours following the stimulus and associated with increased expression of Fos in the TNC in sumatriptan-, but not vehicle-, pretreated rats, suggesting the presence of a state of “latent sensitization” (18,21).
The development of delayed and generalized CA following environmental stress, as seen in the present investigation and in our previous studies (18,19,21), along with enhanced Fos expression, suggests amplification mechanisms that likely include descending facilitation from the rostral ventromedial medulla (RVM) to promote central sensitization of the trigeminal nociceptive pathway. Our previous studies have shown that both facial and hindpaw CA are abolished by inactivation of the RVM with bupivacaine (27). Delayed and generalized CA was produced in animals with sumatriptan-induced latent sensitization following challenge with NO donor or with intravenous (i.v.) injection of CGRP (18,21), provocative stimuli known to elicit migraine headache in susceptible individuals (43). As primary afferent fibers in both the trigeminal and extracephalic regions express the 5-HT1B/D receptor, it is possible that a prior period of triptan exposure could produce sensitization of peripheral afferents innervating the hindpaw. However, triptans have an almost exclusive effect clinically on cephalic pain (44). Cephalic selectivity for triptans was also observed in blocking aversiveness due to dural inflammation, but not hindpaw incision (45). Other studies have also demonstrated differential effects of triptans' trigeminal and spinal primary afferents (46). Whereas naratriptan blocked evoked activity of trigeminal afferents, the activity of spinal afferents remained unchanged (46). Collectively, these observations, together with the delayed onset of CA and increased Fos expression in TNC in response to putative migraine triggers including stress, NO donor or CGRP are consistent with the presence of central amplification mechanisms.
Clinical studies employing advanced neuroimaging techniques provide strong evidence that CSD can evoke migraine (7,47,48), thus acting as a possible endogenous trigger (6,49). Detection of cerebral blood flow changes characteristic of CSD-like events in individuals experiencing migraine headache without aura (50,51) suggests the possibility that “silent” aura may be generated by CSD waves occurring in brain regions that are likely to be asymptomatic (48,52). Studies performed with human neocortical sections showed that even a single CSD event could facilitate excitatory post-synaptic potentials and elicit long-term potentiation, thus contributing to cortical hyperexcitability (53). CSD may also generate headache by activating central trigeminovascular neurons through subcortical pathways that result in disinhibition of sensory neurons within the TNC (54). Meningeal plasma extravasation has been detected by photon-emission tomography during migraine (55).
In the present study, we used a known graded electrical stimulation protocol (24) to determine the stimulation threshold to generate a CSD in rats with triptan-induced latent sensitization. Using this method, we never observed more than one CSD generated once the threshold was reached. Previous studies have employed alternate methods of generating CSD events, including application of either a crystal or a known concentration of KCl onto the dura followed by counting the numbers of generated CSD events. These approaches provide different information that addresses either the electrical threshold required to generate a CSD (present study) or the brain's ability to elicit CSDs in response to a potent inducer (KCl). The simultaneous recording of the EEG waves along with the DC potentials provided confirmation of the presence of CSD, and analysis showed that the fundamental EEG characteristics did not differ among the treatment groups. Finally, the EEG trace provided a means of ensuring that excessive depth of anesthesia was avoided, as this would have been indicated by appearance of burst suppression.
Our data show that a period of prior exposure to sumatriptan markedly and significantly reduced the electrical stimulation threshold required to generate a CSD. This result is consistent with long-lasting increased cortical excitability produced by triptan exposure. Additionally, CSD events generated in sumatriptan-pretreated rats resulted in enhanced expression of Fos in the TNC, consistent with enhanced activation or consequences of activation of the trigeminal pathway. These findings are consistent with previous reports demonstrating that CSD can release pro-inflammatory mediators (56), excite dural afferent fibers (9,52,57), and elicit Fos expression in the outer laminae of the TNC (6). How a period of prior exposure to triptans might elicit enhanced cortical excitability is not known. While sumatriptan has relatively limited blood-brain barrier (BBB) penetration, its brain/plasma partition coefficient of 0.13 suggests that central effects can occur as has been previously noted in humans and in animals (43). Additionally, as triptans are agonists for the 5-HT1B/D receptors, central effects may occur even with low fractional receptor occupancy (58). Thus, it is conceivable that a period of prolonged sumatriptan exposure, as employed here, could lead to consequential levels of the drug within the brain to promote long-lasting central changes underlying cortical excitability.
It should be noted that both latent sensitization in preclinical settings as well as MOH in patients have been observed with drugs that act at multiple receptor classes including, for example, triptans and opioids. We have shown that either opioids or triptans can increase the number of dural afferent cell bodies labeled for excitatory transmitters such as CGRP (18,19,59). Furthermore, prolonged exposure of rats to acetaminophen resulted in increased numbers of CSD events to dural KCl along with increased Fos expression in the TNC, also suggesting increased cortical and trigeminal excitability (60). These observations suggest that triptan-induced cortical excitability likely reflects generalized changes in sensory processing that are not specific to a single mechanism.
In the present study, we used non-noxious environmental stress to produce delayed and generalized (i.e. facial, hindpaw) CA that was accompanied by increased Fos expression in the TNC in rats with triptan-induced latent sensitization. Clinically, stress has been suggested to provoke migraine attacks and may be a risk factor for the chronification of migraine (61). Stress is a powerful signal that elicits activation of numerous central components, including the hypothalamic-pituitary axis, sympathetic responses, and structures of the limbic system, notably the amygdala and thalamus, that may enhance cortical excitability (62–64). Recent studies implicate hypothalamic projections to the lateral and posterior thalamic nuclei in photophobia and migraine induced by emotional reactions (65). While CSD in response to a stressful stimulus has not yet been demonstrated, it is possible to speculate that a stress-induced state of heightened central activity could result in CSD events in individuals with reduced thresholds that could manifest as enhanced trigeminal activation and pain, consistent with increased TNC Fos expression and CA in the present study.
Imaging studies performed on migraineurs with extracephalic CA demonstrated sensitization of dural-specific trigeminovascular neurons in the sensory thalamus (39,40). Labeling studies show extensive projections from dural-sensitive thalamic neurons to cortical sites, including the somatosensory S1 and S2 cortices and the insula (66), indicating direct connection to nociceptive-processing areas. Moreover, it was proposed that disruptions in the oscillatory activity of the motor cortex by chronic or evoked pain attenuates intracortical inhibition as well as inhibition of thalamic relays, which can lead to enhanced cortical excitability (66), thus potentially lowering the threshold for CSD and rendering the brain more susceptible to the effects of migraine “triggers” (42,67). Thus, afferent drive via central trigemino-thalamocortical, especially into motor cortex, pathways may disrupt normal intracortical inhibition thus resulting in cortical hyperexcitability (66). A limitation of the present study is that it was not possible to determine the effects of environmental stress on CSD threshold due to anesthesia.
Further supporting the hypothesis that increased cortical excitability may represent a mechanism of triptan-induced MOH is the observation that a single injection of topiramate abolished the enhanced TNC Fos expression in response to bright light stress or to CSD events as well as stress-induced CA in the present study. Topiramate administration normalized and/or elevated the stimulation thresholds to elicit CSD events. These observations are consistent with the likely mechanisms of action of topiramate to reduce CNS excitability. Topiramate attenuates voltage-gated sodium and calcium channels, blocks α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA)/kainate receptor activity, and enhances the γ-aminobutyric acid type A (GABAA) inhibitory chloride channels (68,69). These properties together are likely to strongly interfere with CSD propagation (20). Accordingly, acute topiramate reduced cortical excitability in human subjects (70) and inhibited the evoked activity of TNC neurons in anesthetized cats (71). Importantly, the inhibition of TNC activity was not due to a direct effect of topiramate on TNC neurons, indicating that topiramate likely blocks excitatory events upstream from the TNC (72). Electrophysiologic studies revealed that topiramate inhibited trigeminovascular activity by blocking kainate receptors in the TNC and the ventroposteromedial thalamic nucleus (69). However, in clinical practice, the administration of topiramate for migraine prophylaxis is given in increasing doses over several weeks in order to mitigate the side effects, especially sedation and cognitive dysfunction. Topiramate is not administered acutely to abort ongoing migraine. In a study designed to explore the activity of acute and long-term administration of common clinically used migraine prophylactic medications, Ayata and colleagues (24) found that administration of topiramate over several weeks was required to elevate stimulation threshold to generate CSD or to attenuate the numbers of CSD events induced by KCl applied to the dura. In that study, acute injection of the same dose of topiramate that was used in the present study did not reduce the number of CSD events to dural KCl, although the CSD propagation speed was reduced, consistent with reduced cortical excitability (24). The effect of acute topiramate on stimulation thresholds to generate CSD was not determined in that study. However, the results of the present investigation are not inconsistent with these observations, since we observed that the electrical stimulation thresholds were not elevated by the same dose of acute topiramate in saline-pretreated rats. Critically, acute topiramate raised the stimulation thresholds only in animals with sumatriptan-induced latent sensitization. It should be noted that the current investigation did not attempt to model prophylaxis of chronic migraine, but rather evaluated the effects of topiramate on CSD threshold and on the downstream consequences of CSD in rats with latent sensitization.
The results of the current investigation suggest that a period of sumatriptan exposure can lead to long-lasting enhancement of mechanisms that promote excitability, including a lowering of thresholds to elicit CSD events. Increased cortical sensitivity to generate CSD events is consistent with clinical observations in MOH patients. Moreover, an enhanced sensitivity of the trigeminal system both to CSD and environmental stress was demonstrated by the elevated Fos expression in the TNC. These results are also consistent with sensitization of the trigeminal pathway in MOH and chronic migraine. These signs of enhanced sensitization were abolished by acute topiramate. A prior period of sustained triptan exposure thus provides an animal model that appears to have translational relevance to triptan-induced MOH and perhaps chronic migraine, and may thus provide a platform for the evaluation of potential prophylactic migraine therapies that are designed to decrease cortical excitability, CSD initiation, and/or activation of sensory neurons within the TNC.
Article highlights
Sumatriptan exposure can lead to latent sensitization of central trigeminovascular neurons, revealed by enhanced trigeminal nucleus caudalis (TNC) Fos expression to cortical spreading depression (CSD) or bright light stress. Triptan exposure reduces the electrical stimulation threshold to elicit CSD, suggesting the possibility of enhanced cortical excitability. Decreased CSD thresholds and enhanced Fos expression are blocked by acute systemic topiramate.
Footnotes
Acknowledgments
The authors thank Janice Oyarzo and Nathan Eyde for their invaluable technical assistance in surgical preparation. We also thank Triza Brion and Naomie Goshima for their assistance in immunohistochemistry. Finally, we express our gratitude to Dr James Alex McGlamery for his assistance in acquiring topiramate for these studies.
Funding
This work was supported by NIH R01 NS069572 (FP). ALG was supported by a Fellowship from the International Headache Society.
Conflict of interest
None declared.