
Abstract
Molecular techniques are increasingly being applied to stained cytology slides for the diagnosis of neoplastic and infectious diseases. Such techniques for the identification of fungi from stained cytology slides have not yet been evaluated. This study aimed to assess the diagnostic accuracy of direct (without nucleic acid isolation) panfungal polymerase chain reaction (PCR) followed by sequencing for identification of fungi and oomycetes on stained cytology slides from dogs, cats, horses, and other species. Thirty-six cases were identified with cytologically identifiable fungi/oomycetes and concurrent identification via fungal culture or immunoassay. Twenty-nine controls were identified with no cytologically or histologically visible organisms and a concurrent negative fungal culture. Direct PCR targeting the internal transcribed spacer region followed by sequencing was performed on one cytology slide from each case and control, and the sensitivity and specificity of the assay were calculated. The sensitivity of the panfungal PCR assay performed on stained cytology slides was 67% overall, 73% excluding cases with oomycetes, and 86% when considering only slides with abundant fungi. The specificity was 62%, which was attributed to amplification of fungal DNA from control slides with no visible fungus and negative culture results. Direct panfungal PCR is capable of providing genus- or species-level identification of fungi from stained cytology slides. Given the potential of panfungal PCR to amplify contaminant fungal DNA, this assay should be performed on slides with visible fungi and interpreted in conjunction with morphologic assessment by a clinical pathologist.
Keywords
Fungal infections have become more prevalent worldwide in recent years, and emerging fungal pathogens surface regularly. 4 Part of the reason for this increase in fungal infections is the increased number of immunosuppressed or immunocompromised individuals and increased lifespans of these patients due to advances in treatment modalities. 12 While many fungal infections are treated similarly, it is clear that certain fungi and organisms that morphologically resemble fungi are inherently resistant to particular antifungal agents. 3,6,8,10,21,22 For example, cutaneous pythiosis and lagenidiosis may require different treatments and have a worse prognosis than cutaneous zygomycosis. 8 Furthermore, several agents of hyalohyphomycosis reportedly exhibit inherent resistance to amphotericin B and itraconazole. 3,25 Accurate identification of the etiologic agent therefore allows for appropriate, targeted treatment. Additionally, identification of fungi is important for determination of pathogenesis, for tracking disease outbreaks, and for identification of novel and emerging fungal pathogens. 10
Cytology is a commonly used, first-line diagnostic tool to investigate lesions on various animal species. 20 It is considered a relatively noninvasive test that provides rapid results to guide treatment and further diagnostics. Fungi can be observed by microscopically examining stained cytology slides, including smears of fine-needle aspirates, tissue impressions, and fluid cytology. Occasionally, organisms display morphologic features characteristic for a specific fungus (eg, the prominent mucopolysaccharide capsule of Cryptococcus spp.). However, as with histologic evaluation, many fungi are difficult or impossible to distinguish even to the genus level based on microscopy alone. 17 Fungi that form hyphae are particularly difficult to classify cytologically, and additional diagnostics are often required in order to determine the genus and species. 9 Additional diagnostics may include fungal culture, immunoassays, and molecular assays.
Many molecular tests are specific for one or a limited number of known fungal organisms. Some methods, such as panfungal qPCR (qualitative polymerase chain reaction) with melt curve analysis and MALDI-TOF MS (matrix assisted laser desorption ionization-time of flight mass spectrometry), are designed to detect a broader range of fungi; however, the results must still be compared to relatively limited databases of fungi. 23,26 On the other hand, panfungal PCR followed by sequencing (PCRsq) can potentially amplify any fungus without prior knowledge of its identity, albeit with an increase in turnaround time. 12 Panfungal PCR amplifies conserved genes that are present in all fungi, including the ribosomal RNA region and its internal transcribed spacer (ITS-1 and ITS-2) regions. 19 Primers targeting these regions produce PCR products with variable sequences that allow for distinction between most fungal species. 19 Panfungal PCR has been tested on several sample types, including formalin-fixed paraffin-embedded (FFPE) tissues from human and veterinary patients. 5,12,15 These studies show that panfungal PCRsq allows identification of fungi to the genus or species level in the majority of cases. To the authors’ knowledge, no studies have been published to date assessing the diagnostic accuracy of panfungal PCRsq performed on cytology slides in either human or veterinary medicine.
Recently, specific PCR assays have been developed for use on cytology slides for the detection of neoplastic and infectious diseases such as mycobacteriosis and leishmaniasis. 14,16,24 Cytology slides may be advantageous compared to FFPE tissues for molecular testing because they can be prepared more rapidly and often without the need for anesthesia; furthermore, the fixation method is more gentle with less degradation of DNA, and there are fewer chances for contamination during processing steps. 1,11,28 This study aimed to evaluate the suitability of stained cytology slides as samples for direct panfungal PCRsq.
Materials and Methods
Selection of Cases and Controls
Medical records from the Texas A&M University Veterinary Medical Teaching Hospital were searched retrospectively from the years 2014 to 2019 for all patients (of any species) where both cytologic examination and fungal culture were performed on the same tissue during a single hospital visit. In the case of dimorphic fungi and oomycetes, commercial immunoassays were also accepted as confirmatory tests given the difficulties of culturing these organisms. If a fungus was identified by both cytology and confirmatory assay, these patients were identified as cases. If a fungus was not identified via cytology or culture and a diagnosis of non-fungal disease was confirmed via histologic examination, these patients were identified as controls.
Archived cytology slides for each case and control were identified from the Texas A&M College of Veterinary Medicine’s Clinical Pathology Laboratory archive. Slides had been archived in drawers without coverslips and had been previously stained with either Diff-Quik (a manual, dip-type, Romanowsky stain) or an automated Wright’s stain. For each case, a representative slide with visible fungal elements was chosen. For each control, the most representative slide was chosen, that is, the slide with the most cells/elements associated with the aspirated tissue.
If a case also had matched FFPE tissues obtained from the same tissue as the cytologic preparation during the same hospital visit, the histology slides and FFPE tissue were also examined microscopically and analyzed with panfungal PCR.
Microscopic Examination and Quantification of Fungi
Each cytology slide (and histology slide, when available) was reviewed for quantity and morphology of fungal elements by a board-certified veterinary clinical pathologist (AM) and veterinary anatomic pathologist (ARH). The following quantification scheme was used for both cytology slides and histology slides: Fungi were quantified in 20 fields at 200× magnification as either rare (<3 yeast or <8 hyphae), occasional (3-10 yeast or 8-20 hyphae), moderate (10-15 yeast or 20-50 hyphae), or abundant (>15 yeast or >50 hyphae). 15 Representative images were taken of each slide, including control cases with no fungi (SPOT Imaging). Following slide review, immersion oil was removed from cytology slides by gentle blotting with a clean Kimwipe (Kimberly-Clark) then dipping in AmeriClear (Cardinal Health, Inc) a histologic clearing solvent. Slides were then air-dried or gently blow-dried and stored in standard 100-slide boxes while awaiting PCR.
Sample Preparation for PCR
After reviewing the chosen cytology slide from each case, the same slide was used to obtain DNA for the PCR reaction. Preparation for direct PCR was performed by scraping a pinpoint amount of material from the cytology slide (from an area where fungi were observed via light microscopy) with a sterile #11 stainless steel scalpel blade directly into a 0.5 ml PCR tube containing already-prepared and portioned PCR reaction mixture, using a similar method as described for colony PCR of cultured fungus. 27 For control slides without any fungi, 2 scraping replicates were performed into separate PCR tubes, both from random locations of the smear.
To prepare FFPE tissues for conventional panfungal PCR, DNA was extracted from 50-µm curls from each FFPE tissue block with the BiOstic FFPE Tissue DNA Isolation Kit (MO BIO Laboratories Inc) following the manufacturer’s protocol.
Conventional PCR and Sequencing
Conventional PCR targeting the fungal ITS-2 region was performed with the following primers on all direct cytology scrapings as well as DNA extracted from FFPE tissues: ITS3-F (5′-GCATCGATGAAGAACGCAGC-3′) and ITS4-R (5′-TCCTCCGCTTATTGATATGC-3′). The total volume of the PCR reaction was 25 µl consisting of 12.5 µL of AccuStart II PCR ToughMix (Quantabio) 0.2 μM of each primer, 1 µL of template or pinpoint scraping of template from cytology slide, and water up to the reaction volume. The reaction mixture was denatured at 95 °C for 3 minutes, followed by 40 cycles of denaturation at 95 °C for 15 seconds, annealing at 59 °C for 15 seconds, and extension at 68 °C for 45 seconds. This was followed by a final extension step for 5 minutes at 68 °C in a T100 Thermal cycler (Bio-RAD Laboratories). Each batch of PCRs included a negative PCR control (template consisting of 1 µl of DNA-free molecular-biology-grade water), a positive PCR control (1 µl of purified Cryptococcus neoformans DNA), and, for direct cytology scrapings, a negative scraping control (scraping of a “blank” glass slide to rule out contamination during this process) and positive scraping control. The positive scraping control was performed using a cytology slide with mats of hyphae from a well-characterized case (concordant culture, histology, cytology, and PCR results) of canine nasal aspergillosis caused by Aspergillus fumigatus.
For control of fungal DNA contamination, all work surfaces and equipment such as pipettes and racks were decontaminated with DNA AWAY (Molecular Bio-Products, Inc) prior to performing experiments and between slide scrapings. PCR master mixes were prepared in Purair PCR Laminar Flow Cabinets (Air Science) that were routinely decontaminated with DNA AWAY and UV lamp. DNA-free filter tips, microcentrifuge tubes, and PCR tubes were used throughout the study.
All PCR products were electrophoretically separated in 2% agarose gel followed by excision of all bands of the appropriate size. The expected product size for the amplified region ranges from approximately 300 to 500 base pairs for fungi and approximately 600 to 700 for oomycetes. DNA was purified from the bands using the E.Z.N.A. Gel Extraction Kit (Omega Bio-tek, Inc) following manufacturer guidelines. Purified DNA was submitted for Sanger sequencing to a commercial laboratory (Eton Bioscience, Inc). Sequence chromatograms were examined, and sequences were manually trimmed to include only high-quality base calls. Sequences were then queried with BLASTn (National Center of Biotechnology Information; www.ncbi.nlm.nih.gov/BLAST) against the NCBI Fungal ITS Targeted Loci database 18 and with the CBS-KNAW pairwise sequence alignment tool (http://www.wi.knaw.nl/Collections/BioloMICSSequences.aspx). Fungi with ≥97% sequence similarity to the queried sequence were reported.
Statistical Analysis
Descriptive statistics were gathered and reported as counts or proportions of cases and/or controls. Additionally, each sample was classified as follows by comparing results of PCRsq to culture or immunoassay results:
True positive (TP)—PCRsq identified a fungus matching the culture or immunoassay identification.
True negative (TN)—PCRsq did not amplify fungal DNA, and the culture or immunoassay was negative. Samples were regarded as a true negative if only Malassezia species DNA was amplified.
False positive (FP)—PCRsq identified a fungus, excluding Malassezia species but the culture or immunoassay was negative.
False negative (FN)—PCRsq did not amplify fungal DNA, but culture or immunoassay identified a fungus, OR PCRsq identified a fungus incompatible with the culture/immunoassay identification. Sequenced fungal DNA was considered incompatible if it did not match the culture/immunoassay identification to at least the genus level.
Sensitivity was determined with the formula TP/TP+FN, specificity with TN/TN+FP, positive predictive value with TP/TP+FP, and negative predictive value with TN/TN+FN. Ninety-five percent confidence intervals for these indices were determined using MedCalc statistical software (MedCalc Software Ltd). A control sample was considered to be FP if either of the 2 scraping replicates was positive for fungal amplification.
Results
Selection of Cases and Controls
A search of medical records and the cytology slide archive yielded 36 cases (Supplemental Table S1) and 29 controls (Supplemental Table S2). The majority of cases and controls were dogs with fewer cats and horses. Two exotic species were included as controls. Nine cases (Cases 5, 6, 12, 16, 21, 28, 29, 30, and 31) had matched FFPE tissues allowing for concurrent histopathologic evaluation. The represented tissue types included a mix of sites that would typically be considered sterile (such as spleen, lymph node, and kidney) and those that are likely to contain normal microbiota including fungal microbiota (such as gastrointestinal tract, cornea, and skin).
Microscopic Examination and Quantification of Fungi
Evaluation of cytology cases revealed a range of yeast and hyphal morphologies (Figs. 1–4) and varying fungal abundances with most cases (22/36) having cytologically abundant organisms and 4 cases having rare organisms. In cases with rare organisms, as little as 2 hyphae (Case 23) or 4 yeast (Case 15) were identified in the entire smear. In no case was more than one type of fungi observed cytologically. Evaluation of cytology controls confirmed a lack of visible fungi on all control slides.
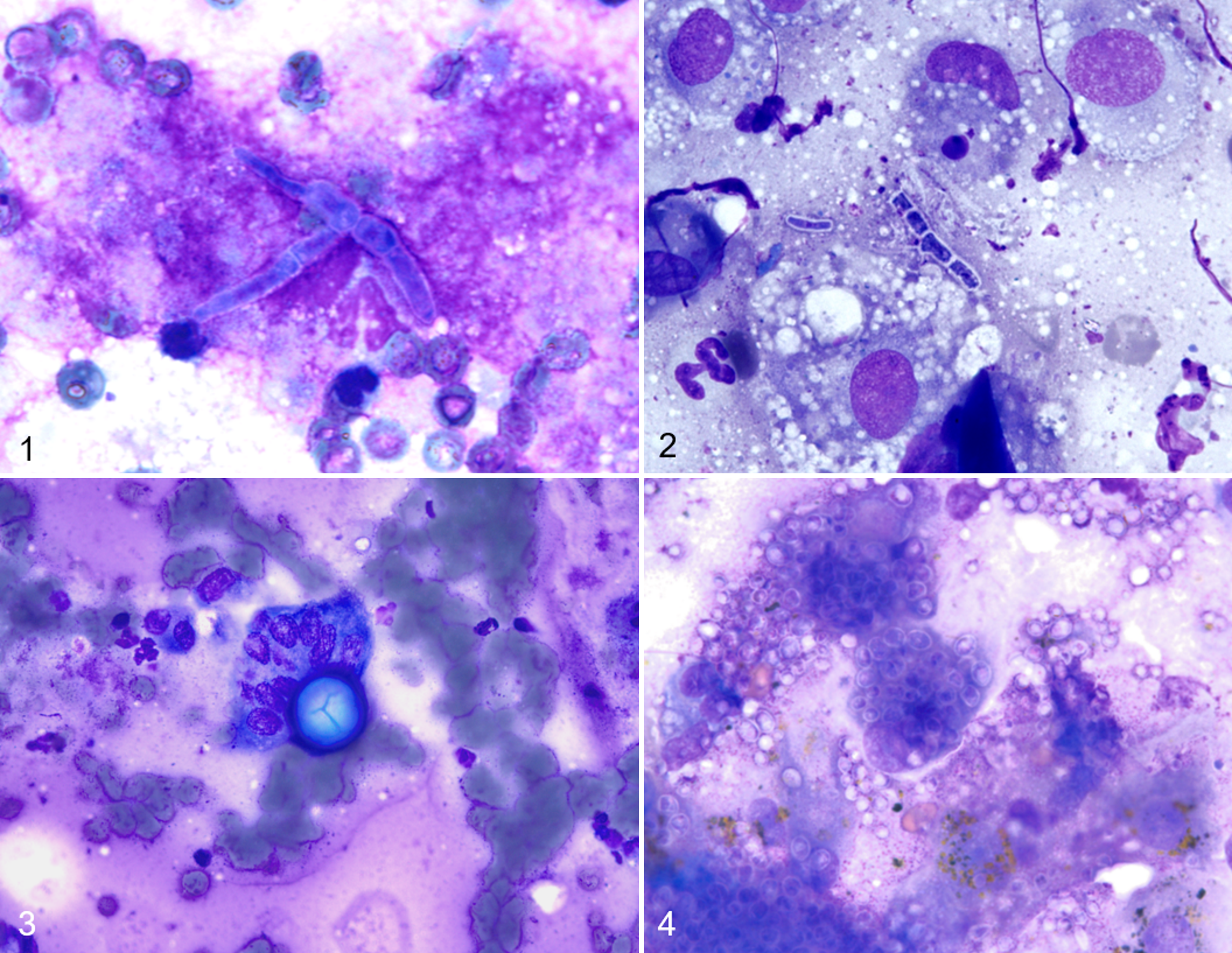
Aspergillus terreus species complex infection, kidney, dog. A single, septate, branched hypha and scattered erythrocytes. Diff-Quik stain.
For the 9 cases with matched histology slides, the histologic fungal morphology matched that of the corresponding cytology slide, and fungal abundance matched in all cases except Case 30, where the cytologic abundance was moderate and the histologic abundance was abundant.
PCR and Sequencing Results
All PCR runs exhibited appropriate negative and positive PCR and scraping control results (no contamination was observed in negative PCR control samples). Analysis of sequencing results revealed that direct panfungal PCRsq identified the same fungus from the cytology slide as the confirmatory method (culture or immunoassay) in 24/36 (67%) cases (Fig. 5). Both yeast and filamentous fungal DNA were successfully amplified, including Aspergillus, Fusarium, Microsporum, and Trichophyton sp., as well as a phaeohyphomycete in the Pleosporaceae family (Case 26). For the 3 cases of pythiosis, no amplification was achieved. Sequencing identified contaminating fungi in 11/29 (38%) of the culture-negative controls (2 controls containing only Malassezia sp. DNA were not included in this count). One of these controls yielded fungal DNA amplification from both scraping replicates (Control 21) while the remainder yielded amplification from only 1 of the 2 replicates. Amplification from either one or both replicates was considered a false positive. Contaminating fungal DNA incompatible with the cytologic appearance or the culture/immunoassay results were amplified in 5/36 (14%) cases (4 cases containing Malassezia spp. as the only contaminant were not included in this count). Malassezia spp. were the predominant contaminants in both case and control slides.
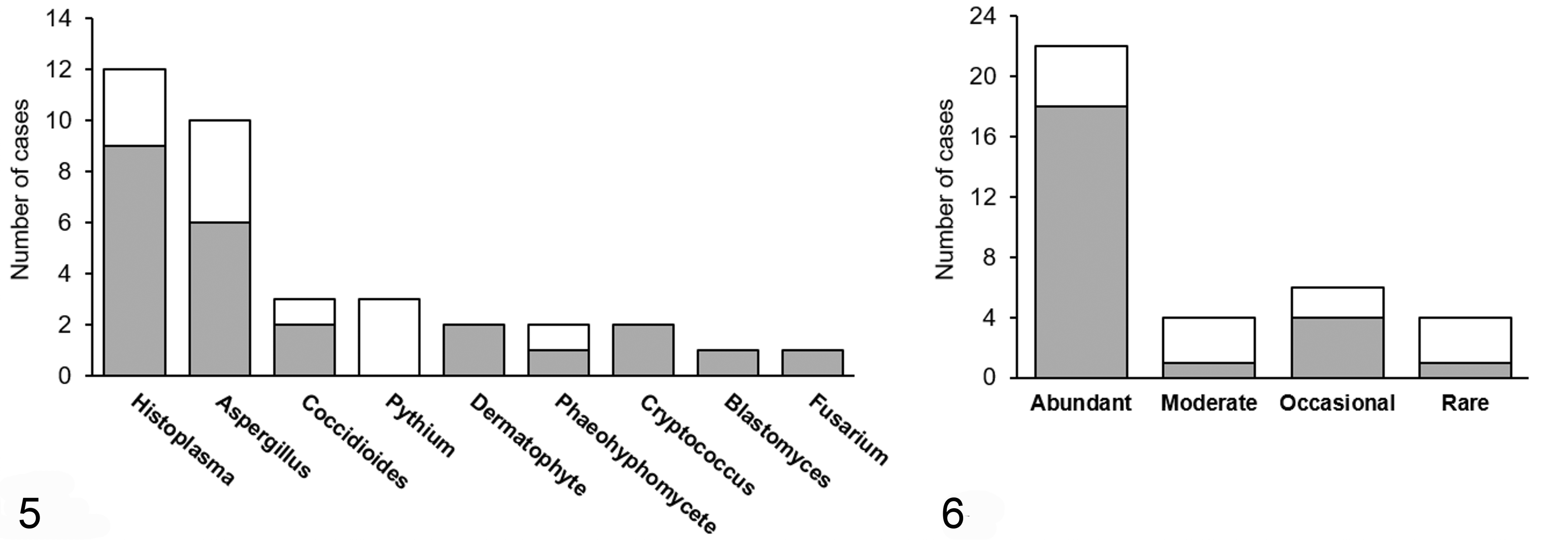
Agreement of cytopathology and PCR with sequencing for identification of fungi. Grey bars indicate the number of cases with agreement between the methods.
Cases with cytologically abundant fungi were more likely to have a positive identification by PCR (Fig. 6). Of the 4 cases with rare organisms, 3 cases exhibited no amplification (2 pythiosis and 1 aspergillosis). Coccidioides sp. was successfully amplified from the remaining slide with rare organisms (Case 15). This slide contained 4 visible spherules consistent with Coccidioides sp., and a single spherule was scraped into the PCR reaction tube.
Regarding the stains used, 14/19 (74%) manually Diff-Quik-stained cases were correctly identified and 10/17 (59%) automatically Wright’s-stained cases were correctly identified. Contaminating fungal DNA was amplified from 3/8 (38%) Diff-Quik-stained controls and 8/21 (38%) Wright’s-stained controls. There was no obvious relationship between type of stain and positive identification of fungi or amplification of contaminant fungi. Likewise, the duration of storage was not overtly related to increased amplification of contaminant fungi (Supplemental Tables S1 and S2). The oldest slide with a positive fungal identification via direct panfungal PCRsq was approximately 5 years old.
Of the 9 cytology cases with matched FFPE tissue available, the PCR and sequencing results yielded concordant positive identification of the fungus in 7 cases (Supplemental Table S3). For Case 21, positive identification was not achieved from either the cytology or the FFPE tissue block despite the presence of abundant, microscopically visible fungi in both sample types. For Case 30, positive identification of Alternaria sp. was achieved only from the FFPE tissue block. Fungi were more abundant histologically than cytologically for this case.
Diagnostic Performance Calculations
Overall sensitivity, specificity, and positive and negative predictive values are displayed in Table 1. The sensitivity of the panfungal PCR assay performed on stained cytology slides was 67% overall, 73% excluding cases with oomycetes, and 86% when considering only cases with abundant fungi. The specificity was 62%, attributed to amplification of fungal DNA from control slides with no cytologically visible fungi and negative culture results.
Performance Indices of Direct Panfungal PCR Followed by Sequencing on Stained Cytology Slides.
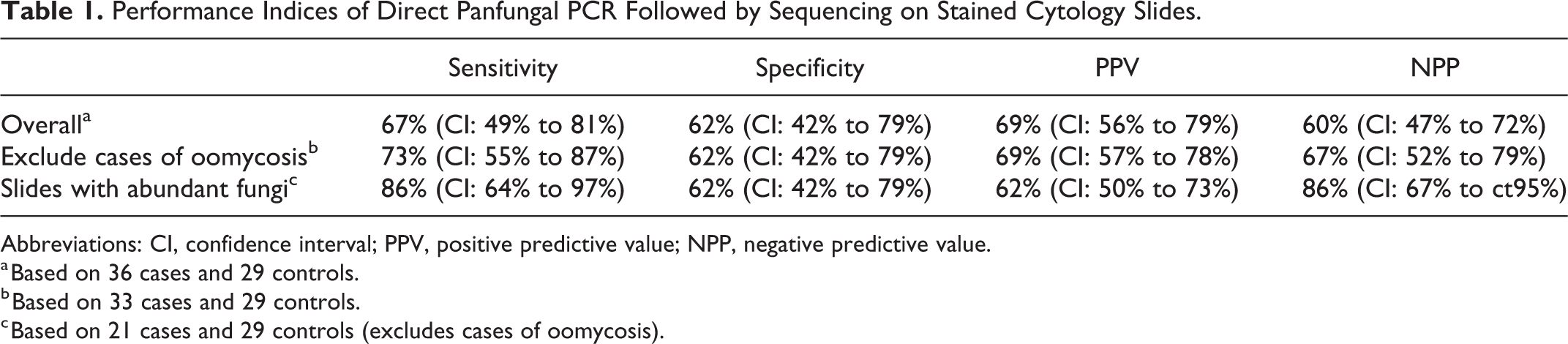
Abbreviations: CI, confidence interval; PPV, positive predictive value; NPP, negative predictive value.
a Based on 36 cases and 29 controls.
b Based on 33 cases and 29 controls.
c Based on 21 cases and 29 controls (excludes cases of oomycosis).
Two controls (Control 17 and 24) yielded poor-quality sequences that could not be used for fungal identification. Overlapping peaks were observed on the chromatograms for these controls, suggesting the presence of multiple DNA templates. These samples were considered to be false positives given that amplification was not expected for the control samples. One case (Case 25) also yielded a poor-quality sequence that did not allow for fungal identification. This result was considered a false negative given that a match to the fungus identified by cytologic examination and immunoassay, Histoplasma capsulatum, was not possible.
Discussion
Cytology slides are a readily obtainable and archivable source of DNA for molecular testing. This study evaluated direct panfungal PCRsq for identification of fungi using stained cytology slides as the source of DNA. Direct panfungal PCRsq positively identified fungi in the majority of stained cytology slides from animals with fungal infections including endemic mycoses (blastomycosis, coccidioidomycosis, and histoplasmosis), cryptococcosis, aspergillosis, fusariosis, phaeohyphomycosis, and dermatophytosis. This assay may be useful in the diagnosis of both common and rare/emerging fungal infections when more ideal sample types cannot be collected for culture or when culture fails. It may also be a useful for diagnosis in cases where faster turnaround times are needed or when culture or immunoassay results are questionable.
The direct PCR method has been shown to successfully amplify yeast, filamentous fungi, and Pythium sp. from culture isolates and to amplify fungi from human corneal swabs without the need for lysis of the fungal cell wall. 13,27,29 This direct method of PCR was chosen for its ability to (1) improve turnaround time, (2) prevent loss of small quantities of DNA to the extraction process, (3) decrease opportunities for sample contamination during the extraction process, and (4) preserve cytology slides for archiving purposes. A comparison of whole-slide DNA extraction and direct PCR was not performed as only a single representative cytology slide was available for most cases.
The low specificity of this assay reflects the ability of panfungal PCR to amplify the DNA of essentially any fungus, including normal inhabitants of the host microbiome and environmental fungi. Previous panfungal PCR studies using FFPE tissues have failed to address this by reporting only sensitivity and not including negative controls 1,15 or by considering identification of common environmental fungi as true negative even though some of these fungi are considered to be opportunistic pathogens. 5 The latter is reasonable in regard to Malassezia species, which have been reported as the most common contaminants in studies assessing panfungal PCR on FFPE tissues and as common contaminants in PCR reagents. 1,2,5 Malassezia are a normal inhabitant of the skin microbiota in both humans and animals and could easily be ruled out as a cause of infection in all cases included in this study based on clinical and cytologic findings. For this reason, when only Malassezia spp. were amplified from controls, these were labeled as true negatives rather than false positives.
It is worth noting that, in this study, contaminants were more commonly amplified from negative control slides containing no visible fungi than from case slides containing pathogenic fungi. A potential explanation for this is that contaminants are less likely to be amplified if a large amount of pathogenic fungal DNA is available on the slide as a template for PCR amplification. 7 Therefore, this assay is expected to be most reliable when paired with cytologic examination and performed on areas of slides with readily visible organisms. Neither the type of stain nor the duration of storage (up to approximately 5 years for this study) exhibited an obvious relationship with contamination or sensitivity, suggesting that several years of archiving does not alter the usefulness of cytology slides as a source of DNA for this assay.
Regarding the 1 case and 2 controls that yielded poor-quality sequences, it is important to realize that panfungal PCR can amplify multiple fungi at one time from the same sample. In some instances, the amplified DNA from multiple fungi cannot be separated electrophoretically for sequencing, resulting in a characteristic chromatogram and poor-quality sequences. Sanger sequencing is therefore not ideal for identifying multiple fungi within a single sample, and modern next-generation sequencing techniques, such as targeted ITS amplicon sequencing, would be better suited to this task. Not unexpectedly, control samples taken from sites expected to have a normal fungal microbiome were more likely to exhibit fungal amplification and poor-quality sequences than control samples collected from sites typically considered to be sterile. The specificity of the assay would likely have been higher if more control samples had been collected from sites considered to be sterile; however, we attempted to include diverse controls that would be similar to cases in this regard.
In 3 cases of oomycosis, no amplification was achieved. While oomycetes are not fungi, they have previously been amplified with the ITS primers used in this study and with direct PCR, so the lack of amplification here was unexpected. 13,15 Two of the 3 cases of oomycosis in this study had rare organisms cytologically (<8 hyphae per twenty 20× objective fields), which may have contributed to amplification failure. Other possible causes include degradation of the DNA by host DNases or the presence of PCR inhibitors unique to oomycosis samples. As direct PCR has been used to amplify Pythium sp. from culture, it seems less likely that components of the oomycete cell wall are inhibiting amplification. 13 Further optimization of this assay for detection of oomycetes is needed, ideally including the use of oomycete-specific primers.
In summary, direct panfungal PCRsq is a method for molecular identification of a broad range of fungi on stained cytology slides. No prior knowledge or suspicion of a particular fungus is required, and results can often be obtained faster than with culture. The sensitivity of the assay is good when fungi are abundant (>15 yeast or >50 hyphae per twenty 20× objective fields) and fair when they are less than abundant. To counteract the low specificity of this assay (ie, amplification of DNA from contaminants), interpretation of the results must be performed with concurrent examination of the cytology slide by a board-certified clinical pathologist. Cytologic examination would allow for some sequenced fungal DNA contaminants to be interpreted as a negative result based on fungal morphology. This assay would be most useful in instances where it is not possible to obtain tissues for culture or histologic evaluation but a cytology slide with visible fungi is available for testing.
Supplemental Material
Supplemental Material, sj-pdf-1-vet-10.1177_0300985821991562 - Diagnostic Accuracy of a Direct Panfungal Polymerase Chain Reaction Assay Performed on Stained Cytology Slides
Supplemental Material, sj-pdf-1-vet-10.1177_0300985821991562 for Diagnostic Accuracy of a Direct Panfungal Polymerase Chain Reaction Assay Performed on Stained Cytology Slides by Alexandra N. Myers, Unity Jeffery, Zachary G. Seyler, Sara D. Lawhon and Aline Rodrigues Hoffmann in Veterinary Pathology
Footnotes
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: Alexandra N. Myers was supported by a postdoctoral fellowship from the National Institutes of Health under award number T32OD011083.
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.