
Abstract
The second internal transcribed spacer (ITS2) of ribosomal DNA (rDNA) is used as a genetic marker to identify trichostrongylid nematodes. However, it is often difficult to amplify by polymerase chain reaction (PCR) the ITS2 rDNA of a single trichostrongylid nematode larva or egg. A nested PCR (nPCR) assay was, therefore, developed to amplify the ITS2 from individual trichostrongylid nematode larvae. The results show that the ITS2 rDNA of a significantly greater proportion of individual larvae was amplified using nPCR compared with a standard PCR. There was also no need to column-purify the genomic DNA before nPCR, which is more time and cost effective for studies involving large sample sizes. The amplicons produced from the secondary phase of the nPCR were subjected to single-strand conformation polymorphism analyses and DNA sequencing to confirm the species identity of the larvae used in the current study as Ostertagia gruehneri. The nPCR assay was also used to amplify the ITS2 from individual trichostrongylid eggs. The ability to amplify the ITS2 rDNA from large numbers of individual nematode eggs and larvae has important implications for diagnostic testing and for conducting epidemiological studies on these parasites of veterinary importance.
Keywords
Trichostrongylid nematodes are parasites of major economic importance throughout the world because of their effect on the health of their hosts. Trichostrongyle infections in domestic animals (e.g., sheep, goats, cattle, and pigs) result in reduced wool, meat, and milk production; losses in weight and fertility; sickness; and death. 3 Different trichostrongylid species may differ in their susceptibility to specific chemotherapeutic agents (anthelmintics) 19 or in their effects on hosts. 1,3,24 For example, Ostertagia gruehneri and Marshallagia marshalli are common abomasal parasites of reindeer and caribou (Rangifer tarandus); however, reduced fecundity in reindeer only occurs in response to infection with O. gruehneri and not with M. marshalli. 1,24 Mixed-species infections are also common in wild and domestic ruminants. 15 It is not always possible to reliably and morphologically distinguish eggs of different nematodes within the family Trichostrongylidae at the species level, and sometimes not at the genus level. 12,18,22 A common practice is to culture eggs through to third-stage larvae for identification, which is a time-consuming process (7–14 days) that requires trained personnel. 18 Larvae can be identified morphologically to the genus level but rarely to the species level. 12,18,22
Comparison of the effectiveness of the nested polymerase chain reaction (nPCR) for the amplification of the second internal transcribed spacer ribosomal DNA of individual trichostrongylid nematode larvae subjected to different methods of genomic DNA (gDNA) preparation.*

I = gDNA extracted and column-purified using a commercial kit a ; II = gDNA isolated using an extraction buffer but without column purification; PCR = polymerase chain reaction.
PCR using primers NC13aF and NC2.
PCR using primers NC14-Fn and NC2-Rn and purified products from the primary PCR as template.
Accurate identification of individual nematode eggs and larvae to species is important for the diagnosis of infections, for parasite control, and for population genetics and experimental studies. Several different polymerase chain reaction (PCR)—based assays (e.g., PCR, PCR-RFLP [PCR—restriction fragment length polymorphism], and PCR-SSCP [PCR—single-strand conformation polymorphism]) have been developed to distinguish among adults of different species of trichostrongylid nematodes using the second internal transcribed spacer (ITS2) of the ribosomal DNA (rDNA) as the genetic marker. 3,6,8,20
A crucial step in the effective use of PCR-based assays in diagnosing nematode infections and in population genetic studies is the adequate recovery of genomic DNA (gDNA) from individual eggs or larvae. Several methods have been used to extract the gDNA from a single trichostrongylid egg or larva, 9,14,21,23 or from a pool of eggs, 13,14,25,27 and from individual eggs or larvae of other bursate nematodes (e.g., metastrongyles). 2,5 Although there are a number of commercially available kits that can be used to isolate and purify nematode gDNA, the ability to successfully amplify gDNA from small sample volumes (e.g., single nematode egg or larva) has potential problems. For example, some components within fecal matter (e.g., polysaccharides, humic acid, and fungal contaminants) can inhibit PCR assays. The type of preservative used can also interfere with PCR. 13 Amplification success of gDNA from Ostertagia ostertagi eggs varies depending on the commercial DNA extraction kit used and on the number of eggs used in a sample to extract the gDNA. 14 In 1 experiment, 4 different types of extraction kits were each used to extract gDNA from 5 samples comprising a single O. ostertagi egg; however, amplicons of high yield were only detected in 3 of 20 samples (15%), all of which, were prepared using the same kit. 14 Recently, the authors of the current study encountered difficulties in successfully amplifying the ITS2 rDNA from individual nematode larvae from a variety of sources using a standard PCR. 9 In the current study, the establishment of an effective nested PCR (nPCR) assay to amplify the ITS2 rDNA from individual larvae is described.
The nPCR assay was tested and compared with a standard PCR 9 using larval trichostrongylid nematodes (O. gruehneri) recovered from the fecal samples of 3 experimentally infected animals, 2 reindeer and 1 muskox (Ovibos moschatus). Eggs were isolated using salt flotation and then maintained in distilled water in a petri dish for up to 72 hr to allow larvae to hatch. All larvae from the same host were transferred using a micropipette to a microcentrifuge tube containing phosphate buffered saline (PBS) and stored at −70°C until required for molecular analyses.
Pooled larval samples were thawed and placed into separate sterile Petri dishes containing PBS. Larvae were transferred individually into separate 1.5-ml tubes using a micropipette. Light microscopy was used to verify the presence of a single larva in each tube. Initially, gDNA was isolated from 27 individual larvae (9 from the muskox and 18 from a single reindeer) using a small-scale sodium dodecyl sulfate-proteinase K extraction procedure. 9 The gDNA was then column-purified using a commercial kit a according to the manufacturer's conditions, except that samples were eluted in 30 μl of NANOpure water, rather than the recommended 50 μl of sterile water or TE (Tris-ethylenediamine tetra-acetic acid [EDTA]) buffer. Purification of adult nematode gDNA using this approach has been very effective in amplification by PCR of the ITS2 rDNA. 3,6,9,20 The column-purified larval gDNA of the 27 samples was subjected to PCR using primers NC1 (5′-ACGTCTGGTTCAGGGTTGTT-3′) and NC2 (5′-TTAGTTTCTTTTCCTCCGCT-3′). 9 These primers amplify the ITS2 rDNA and flanking sequences of the 5.8S and 28S ribosomal RNA (rRNA) genes of adult worms of many species of bursate nematode 3,9 and of individual larvae and eggs of some trichostrongylid nematodes. 9 The PCR assays were conducted in 25-μl volumes containing 2.5 mM MgCl2, 62.5 μM of each deoxyribonucleotide triphosphate (dNTP), 20 pmol of each primer, 0.6 U Taq polymerase, and 2–5 μl of gDNA. Negative (i.e., no DNA) controls were included in each set of reactions. The PCR conditions used were 95°C for 5 min, 35 cycles of 95°C for 60 sec, 52°C for 60 sec, and 72°C for 60 sec, and a final cycle of 72°C for 5 min. Of the 27 larval samples tested, a single band (≈325 base pair [bp]) was detectable for only 5 (19%) amplicons (3 larvae from muskox and 2 larvae from reindeer) on 1.5% agarose—TBE (65 mM Tris—HCl, 22.5 mM boric acid, 1.25 mM EDTA; pH 9) gels stained with SYBRsafe b and visualized with ultraviolet transillumination. No bands were detected on agarose gels of the negative control samples. Identical results were obtained when the PCR assays were repeated a second time.
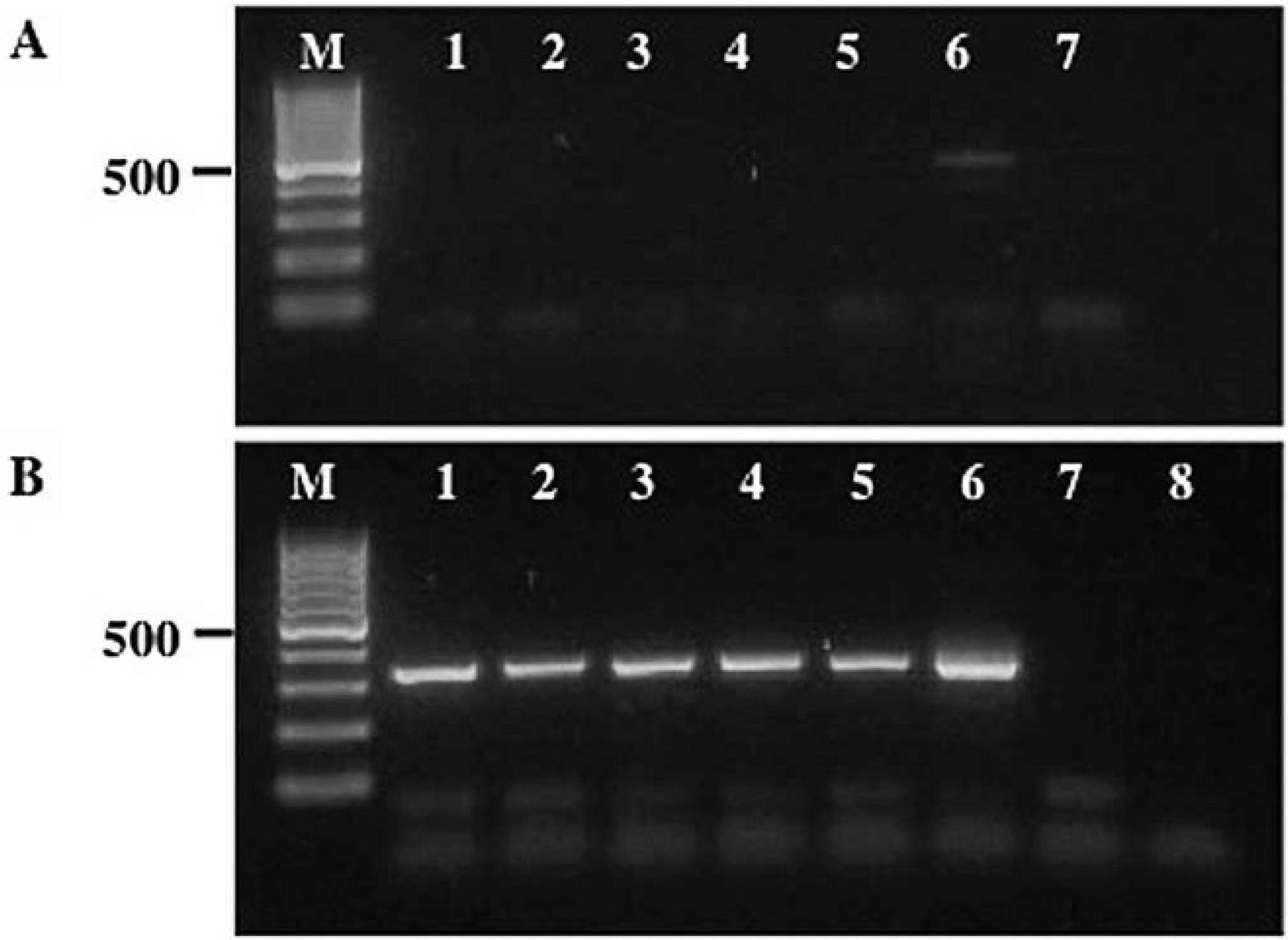
Agarose gels showing the nested polymerase chain reaction (nPCR) amplification of the second internal transcribed spacer (ITS2) ribosomal DNA for 6 representative Ostertagia gruehneri larvae.
Given the low proportion of larvae that successfully amplified, a nPCR assay was designed to amplify the ITS2 rDNA of individual larvae. In addition, a different gDNA extraction method was used, similar to that used for individual Trichinella larvae, 26 in which there was no purification step. Another 105 larvae from the 3 experimentally infected hosts (Table 1) were each transferred to a separate 1.5-ml microcentrifuge tube to which 15 μl of extraction buffer was added. The extraction buffer (500 ml) consisted of 50 μl PCR 10x buffer, 100 μl MgCl2 (12 mg/ml), and 350 μl DNAse-free H2O. Samples were then placed at 90°C for 15 min; after which, 5 μl of proteinase K (500 μg/ml) was added to each tube. Samples were vortexed and then incubated at 58°C for 12–24 hr. Following incubation, samples were heated at 90°C for 15 min and then centrifuged at 10,000 x g for 5 min. The unpurified gDNA in these samples was used as the template in the nPCR amplification of the ITS2 rDNA.
In the first phase of the nPCR, the ITS2 rDNA of all samples were amplified using primers NC13aF (5′-ATC-GATGAAAAACGCAGC-3′) and NC2. Primer NC13aF has the same sequence as primer NC13 3 except for 1 nucleotide position (G→A; position 10). The PCR assays were conducted in 25-μl volumes containing the same concentrations of MgCl2, dNTPs, and Taq polymerase as in the previous experiment, except that 5 μl of gDNA was used as the DNA template. As before, no-gDNA controls were also included in each set of reactions. The PCR conditions used were 95°C for 5 min, 30 cycles of 95°C for 60 sec, 50°C for 60 sec, and 72°C for 60 sec, and a final cycle of 72°C for 5 min. The results of the PCR assays (Table 1) showed that only 36 (34%) of the samples had a single band (≈450 bp) on a 1.5% agarose—TBE gel. One sample (lane 6, Fig. 1A) also had a second weaker band (<400 bp) that probably represented an amplification artifact. The same PCR assays were also conducted on the column-purified gDNA samples of 27 larvae; however, no bands were detected in the amplicons of any of these samples. No bands were detected for the negative control samples.
In the second phase of the nPCR, the products of all nematode samples generated from phase 1 (irrespective of whether or not they were positive on agarose gels) and all the negative control samples were column-purified using commercial PCR purification kits. c These samples were then subjected to PCR using 2 new primers, NC14-Fn (5′-GAACGCATAGCGCCGTTGGGTT-3′) and NC2-Rn (5′-TGATATGCTTAAGTTCAGCGGG-3′), internal to primers NC13aF and NC2. The authors designed (and tested) the 2 new primers based on comparisons of DNA sequences of the 5.8S and 28S rDNA genes from a range of bursate nematodes, including trichostrongyles, strongyles, and metastrongyles. 4 The PCR assays were conducted as in the first phase of the experiments, except that 2 μl of the column-purified products from phase 1 were used as the DNA templates and the annealing temperature was raised to 58°C. An additional negative control was also included in each set of PCR assays. The results revealed that the gDNA of 87% of individual larvae was successfully amplified (Table 1), as evident by the single band (≈350 bp) on the agarose—TBE gels (Fig. 1B). Similarly, for the 27 column-purified gDNA samples, 20 (74%) produced a single band on agarose gels. No bands were detected on agarose gels for the different sets of negative control samples.
Given that the nematode larvae used in these experiments were derived from experimental infections, their species identity was verified using DNA sequencing. The SSCP analyses were used initially on all amplicons derived from the second phase of the nPCR to screen for genetic variation. Samples were prepared for SSCP using methods previously described. 10,17 Samples (5 μl) were loaded into precast gels d that had been placed in a horizontal electrophoresis apparatus d containing 1x TE buffer. A temperature-controlled, circulating water bath connected to the electrophoretic apparatus maintained a constant temperature of 7.4°C throughout the 18 hr that samples were subjected to electrophoresis. Gels were stained for 30 min with SYBR Gold, b then rinsed in distilled water and photographed using an imaging system. e There were 6 different SSCP banding patterns (i.e., profiles) among samples; however, all samples shared 3 bands in common. Amplicons (n = 25) representing each SSCP profile type were subjected to automated DNA sequencing using primers NC14-Fn and NC2-Rn in separate reactions. The ITS2 rDNA sequences (238 bp) obtained were identical to one another, except at one nucleotide position (position 108), where individuals had either an adenosine or a guanine or both nucleotides (sequences deposited in GenBank under accessions FN645446, FN645447, and FN645448), and were consistent with that of O. gruehneri based on comparisons with those of known reference specimens (accession AJ400716). 7
An inability to amplify the gDNA from a single larva or egg represents a major constraint in the species-level identification of bursate nematodes from fecal samples. In the current study, nPCR was used to amplify the ITS2 rDNA from gDNA of larvae that hatched from eggs excreted in the feces of experimentally infected reindeer and muskoxen. The results of the SSCP analyses and DNA sequencing confirmed that the larvae tested were consistent with the experimental infections of O. gruehneri and that no other trichostrongylid nematodes were present. A standard PCR assay used to amplify the ITS2 of adult bursate nematodes 9 was of limited success in the amplification of the same DNA target from individual larvae. Based on the results of the nPCR assay, it is highly probable that the standard PCR used in the current study, amplified the ITS2 of many of the larvae; however, the yield in the amplicons was too low to be detected using agarose gel electrophoresis. In contrast, the ITS2 rDNA was successfully amplified from a high proportion (>85%) of individual nematode larvae using the nPCR assay. Furthermore, there was no need to column-purify the gDNA extracted from individual larvae before nPCR, which represents a more time- and cost-effective approach for diagnostic and population genetic studies involving the analysis of large numbers of individual larvae. However, greater amplification success was achieved following purification of the amplicons derived from first phase of the nPCR (using spin columns or ExoSap-IT g ) rather than using the unpurified amplicons as the DNA template (data not shown). The nPCR assay has subsequently been used to amplify the ITS2 rDNA from single eggs of 3 other genera of trichostrongylid nematodes (i.e., Nematodirus [n = 12], Marshallagia [n = 2], and Teladorsagia [n = 1]) collected from naturally infected hosts. The gDNA of 17 individual eggs was isolated using the extraction buffer—proteinase K method described and subjected to nPCR. Amplicons were detectable on 1.5% agarose—TBE gels for only 2 (12%) of the 17 eggs from phase 1, compared with 15 (88%) eggs from phase 2 of the nPCR. The amplification success of the nPCR for individual eggs was, therefore, similar to that obtained for the O. gruehneri larvae (Table 1). The primer pair NC14-Fn and NC2-Rn also has the potential to be used in the nPCR of other veterinary and medically important bursate nematodes (e.g., hookworms, lungworms, and nodule worms).
The nPCR assay developed in the present study can be used to amplify the ITS2 rDNA (an important DNA target used for species identification of bursate nematodes 3 ) from minute amounts of nematode DNA and to reduce the potential effects of PCR inhibitors and, hence, allowing SSCP analyses and/or DNA sequencing to be conducted on the amplicons produced. The PCR-SSCP of the ITS2 rDNA is a very useful molecular approach for displaying mutational changes among individuals within populations and genetic differences among nematode species. 11,16 The broad applicability of the nPCR assay to amplify the ITS2 rDNA of any species of bursate nematode, particularly those of socioeconomic importance, will, therefore, have important implications in the diagnosis of parasitic nematode infections.
Acknowledgements The authors thank Dr. Florence Huby (Canadian Food Inspection Agency, Saskatoon, Canada) for her valuable suggestions on this work, and the assistance of Dean Brown in the laboratory (of SJK) in Calgary, Canada. The authors also thank the anonymous reviewers for their comments on the manuscript. This research was supported by grants from the National Science and Engineering Research Council of Canada (to NBC, KAS, and SJK), the Canadian Foundation for innovation (to NBC), and the Alberta Ingenuity Fund (to BH and SJK).
Footnotes
a.
Wizard® DNA CleanUp Kit, Promega Corp., Madison, WI.
b.
Invitrogen Corp., Carlsbad, CA.
c.
MinElute®, Qiagen Inc., Valencia, CA.
d.
GMA™ Wide Mini S gels, SEA2000™; Elchrom Scientific, Cham, Switzerland.
e.
BioDoc-It®, UVP LLC, Upland, CA.
f.
Fermentas Canada Inc., Burlington, Ontario, Canada.
g.
GE Healthcare, Piscataway, NJ.