
Abstract
Equid alphaherpesvirus 1 (EHV-1) causes myeloencephalopathy in horses and occasionally in non-equid species. Although mouse models have been developed to understand EHV-1 pathogenesis, few EHV-1 strains have been identified as highly neurovirulent to mice. The aim of this study was to evaluate the pathogenesis of 2 neurovirulent EHV-1 strains in mice, and to characterize the inflammatory cells and expression of chemokines and the apoptosis marker caspase-3 in the brain of infected mice. C57BL/6J mice were inoculated intranasally with EHV-1 strains A4/72 or A9/92 and evaluated on 1, 2, and 3 days post inoculation (DPI). EHV-1-infected mice showed severe neurological signs at 3 DPI. Ultrastructural analysis revealed numerous viral nucleocapsids and fewer enveloped virions within degenerated and necrotic neurons and in the surrounding neuropil. Histologically, at 3 DPI, there was severe diffuse neuronal degeneration and liquefactive necrosis, prominent microgliosis, and perivascular cuffing composed of CD3+ cells (T cells) and Iba-1+ cells (macrophages), mainly in the olfactory bulb and ventral portions of the brain. In these areas, moderate numbers of neuroglial cells expressed CCL5 and CCL2 chemokines. Numerous neurons, including those in less affected areas, were immunolabeled for cleaved caspase-3. In conclusion, neurovirulent EHV-1 strains induced a fulminant necrotizing lymphohistiocytic meningoencephalitis in mice, with microgliosis and expression of chemokines and caspase-3. This model will be useful for understanding the mechanisms underlying the extensive neuropathology induced by these viral infections.
Equid alphaherpesvirus 1 (EHV-1), a member of the Alphaherpesvirinae subfamily, causes several clinical manifestations in its natural host, the horse, including a neurological disorder. 40 Although EHV-1 classically induces a respiratory disease, it is also neurotropic similar to other alphaherpesviruses, such as human alphaherpesvirus 1 (herpes simplex virus) 28 and bovine alphaherpesvirus type 5. 32 The neurotropism manifests as outbreaks of myeloencephalopathy in horses. 42 In horses, EHV-1 initially infects nasal epithelial cells and then is carried to the regional lymph nodes where it infects lymphocytes and monocytes, establishing a cell-associated viremia. 29 Infected leukocytes can reach the central nervous system (CNS), and then EHV-1 infects endothelial cells stimulating a local immune response resulting in thrombosis, inflammation, and necrosis. 42 Besides horses, EHV-1 has also been implicated as a cause of fatal meningoencephalitis with neuronal necrosis in other non-equid species, such as black bears, Thomson’s gazelles, and others. 1,16,45,53
Several studies have developed mouse models of EHV-1 infection. 4,5,18 However, some of these EHV-1 strains have a strong tropism for the respiratory system, and few studies have demonstrated fatal CNS infection induced by EHV-1 in adult mice when inoculated intranasally. To date, one of the most neurotropic EHV-1 strains reported for mice is the Ab4 strain, which causes 50% mortality at 3 days post inoculation (DPI) at a high inoculation dose. 15 In contrast, our research group have previously identified 2 EHV-1 strains (A4/72 and A9/92) that were extremely neurovirulent to different mouse strains and hamsters. 33,36 A4/72 and A9/92 EHV-1 strains induced a severe neurological disease with 100% mortality within 3 to 4 DPI, demonstrating a high neurotropism when compared to other EHV-1 strains. 36
Once in the CNS, herpesviruses elicit an innate immune response by CNS resident cells, with an increase of several chemokines and cytokines. These inflammatory mediators are responsible for the influx of blood-derived leukocytes that may control viral infection, resulting in encephalitis. 6 Several cytokines such as interferon (IFN)-γ, tumor necrosis factor (TNF)-α, interleukin (IL)-1β, IL-6, IL-12, and chemokines CCL5, CXCL9, and CXCL10, among others, have been demonstrated to play a role during herpesviral infection. 3,47,52 In EHV-1 infection, it was demonstrated that the chemokine CCL2 was upregulated in equine airway epithelial cells, 49,55 as well as CCL5 in equine endothelial cells. 25 Moreover, the CCL3 chemokine was responsible for recruiting neutrophils and macrophages to the lungs of EHV-1-infected mice. 51 On the other hand, inflammatory cells and their mediators can indirectly cause extensive damage to the neuroparenchyma. 9 Therefore, the aim of the present study in mice was to correlate the histologic and ultrastructural lesions induced by infection with highly neurovirulent EHV-1 strains with the expression of inflammatory and apoptosis markers.
Material and Methods
Virus
Two different strains of EHV-1 (A4/72 and A9/92), which were originally isolated from aborted equine fetuses, were used in the present study. When inoculated intranasally, these strains were highly neuropathogenic to different mouse strains 36 and hamsters. 33 Although these EHV-1 strains demonstrated a potential neuropathogenicity in animal models, they were previously classified as being non-neuropathogenic according to genetic sequencing of ORF30, which encodes the catalytic subunit of DNA polymerase. 37,39 Viruses were propagated in E. Derm cells (CCL-57, ATCC) and then maintained in Eagle’s minimal essential medium (EMEM) supplemented with 10% fetal calf serum at 37 °C in a humidified atmosphere of 5% CO2.
Mice and Sampling
Thirty-six, 5- to 6-week-old, male, inbred C57BL/6J mice were obtained from the animal facility of the Department of Pathology, School of Veterinary Medicine and Animal Science (FMVZ), University of Sao Paulo (USP), Sao Paulo, Brazil. Mice were housed in individually ventilated cages (Alesco Indústria e Comércio) with pine shavings for bedding, and a controlled room temperature of 22 ± 2 °C, with an air relative humidity of 45% to 65%. They had unrestricted access to filtered and autoclaved water and to irradiated commercial diet (Nuvilab). The room was kept on a 12-hour light/12-hour dark cycle at artificial light. All experimental procedures were approved and carried out according to the Ethics Committee for the Use of Animals of FMVZ, USP (Protocol Number 1174051216).
Thirty-two mice were anesthetized with sevoflurane and intranasally inoculated with 30 µl of EMEM containing 105 TCID50 of each virus (Suppl. Table S1). For histologic and immunohistochemical analysis, 4 EHV-1-infected mice of each viral strain were then euthanized in each time point, including 1, 2, and 3 DPI. Moreover, 4 EHV-1-infected mice were euthanized at 3 DPI per viral strain, and the brains were analyzed using electron microscopy. Four control animals were inoculated with 30 µl of EMEM identically as described for the experimental group, and these mice were euthanized at 3 DPI. Mice were haphazardly assigned to the experimental groups. The body weight of each mouse, including mice from the control group, was recorded daily. In addition, mice were monitored daily for neurological signs such as seizures, crouching in corners, hindlimb paralysis, and recumbency, as well as for other signs such as hunched posture and ruffled hair.
Electron Microscopy
The brain of 4 mice from each group inoculated with the EHV-1 strains was collected at 3 DPI, finely trimmed, and fixed for 24 hours in a paraformaldehyde-glutaraldehyde solution (Karnovsky’s fixative). After fixation, the olfactory bulb (OB) and frontal cortex samples were placed in 0.1 M phosphate buffer, pH 7.25, and then processed for electron microscopy (EM). After washing several times in 0.1 M phosphate buffer, the tissue samples were post-fixed in 1% osmium tetroxide in buffer for 1 hour. The tissue samples were washed 4 times in deionized water prior to en bloc staining with 0.5% uranyl acetate. Vials of tissue were kept in the dark for 1 hour. The tissue samples were then washed several times in deionized water before dehydration in ethanol and cleared in 2 changes of acetone and 2 changes of propylene oxide. The tissue samples were infiltrated with 2:1, 1:1, and 1:2 mixtures of propylene oxide and Mollenhauer’s Epon-Araldite 35 plastic mixture for 3 hours, overnight, and overnight again, respectively. Then samples were further infiltrated with 2 changes of 100% Epon-Araldite plastic mixture for 2 hours each before embedding the samples in fresh plastic using flat embedding molds. The embedded samples were polymerized in a 70 to 80 °C oven overnight.
Sections of 1 µm from the polymerized blocks were obtained using a Reichert Ultracut S ultramicrotome (Leica Microsystems). Sections were placed on glass slides and stained with 1% toluidine blue O in 1% sodium borate. The stained sections were evaluated and areas of interest were chosen before trimming the corresponding block face for thin sectioning. Sections of 60 to 70 nm thickness were obtained and placed on 200-mesh copper grids. The grids were post stained with Reynold’s lead citrate. Grids were viewed with a JEOL JEM-1011 transmission electron microscope (JEOL) at varying magnifications using an accelerating voltage of 100 kV. Images were captured with an XR80M wide-angle multi-discipline mid-mount CCD camera (AMT).
Histopathology
The entire brain and nasal cavity of 4 EHV-1-infected mice per group were collected at 1, 2, and 3 DPI and fixed in formalin for 24 to 48 hours. Four control mice brains collected at 3 DPI were used as controls. The nasal cavity with the OB was decalcified in 14% EDTA, pH 7.0, prior to tissue processing. The tissues were processed routinely for histology and embedded in paraffin.
Sections of 5 µm were prepared and stained with hematoxylin-eosin (HE) to assess histologic lesions. Coronal sections of different CNS regions were evaluated, including the OB, piriform cortex, septal striatum, septal diencephalon, caudal diencephalon (featuring hippocampus and thalamus), rostral mesencephalon (featuring caudal hippocampus), rostral cerebellum with cerebellar peduncles, and spinal cord. The nature of histologic lesions, such as inflammatory or degenerative/necrotic lesions, as well as intensity and distribution of these lesions throughout the CNS were evaluated and described.
Immunohistochemistry
Immunohistochemistry (IHC) for detection of chemokines CCL5 and CCL2, the apoptosis marker (activated caspase-3), T lymphocytes (CD3), and macrophages/microglia (Iba-1; ionized calcium-binding adaptor protein-1), 2,23 were performed on the CNS of control mice and mice infected with 2 different strains of EHV-1 (A4/72 and A9/92) at 3 DPI. For IHC, the paraffin-embedded tissues were sectioned at 5 µm, mounted on slides, dewaxed, and rehydrated. Then, endogenous peroxidase activity was inhibited with a solution of peroxide hydrogen at 3% at room temperature for 5 minutes. Similar to that described in other studies, 20,34 for the detection of CCL2 and CCL5, slides were subjected to antigen retrieval with Borg Decloaker high pH (Biocare Medical) for 15 minutes at 120 °C in a decloaking chamber. Sections were blocked with Power Block (Biogenex) for 10 minutes at room temperature and incubated with anti-CCL2 (AF-479-NA) and CCL5 (AF-478-MR) primary antibodies (R&D Systems) for 1 hour at room temperature at 1:800 and 1:200 dilution, respectively. For the detection of T lymphocytes (CD3) and macrophages/microglia (Iba-1), sections were subjected to antigen retrieval with citrate buffer, pH 6.0, for 15 minutes at 110 °C in a decloaking chamber. The slides were blocked as previously described and incubated for 1 hour at room temperature with anti-CD3 (A0452, Dako Agilent) at 1:1000 and anti-Iba-1 (019-19731, Wako Chemicals) at 1:8000. For detection of activated caspase-3, antigen retrieval was performed using Target Retrieval Solution, pH 9 (Dako Agilent), for 15 minutes at 110 °C in a decloaking chamber and incubated with anti-cleaved-caspase-3 (CP229C, Biocare Medical), which detects a large fragment of activated caspase-3, at 1:16,000 for 1 hour at room temperature. After incubation with primary antibodies, slides were incubated with a secondary biotinylated anti-goat or anti-rabbit immunoglobulin (Vector Labs), followed by a peroxidase-conjugated streptavidin label (4plus Streptavidin HRP Label, Biocare Medical). The binding between antigens and antibodies was visualized using 3,3′-diaminobenzidine substrate (Vector Laboratories). Sections of spleen and lymph nodes from naïve C57BL/6J without histologic lesions were used as positive controls. As negative control, sections of the brain from naïve C57BL/6J mice were used. As an additional negative control, primary antibodies were replaced by non-immune homologous sera.
The pattern and distribution in the brain of the immunostaining for chemokines CCL5 and CCL2, T lymphocytes (CD3), macrophages/microglia (Iba-1), and apoptotic marker (activated caspase-3) in both EHV-1-infected mice and controls were evaluated and described.
Results
Outcome of EHV-1 Infection in Mice
All inbred C57BL/6J mice showed severe neurological disease following EHV-1 infection, especially at 3 DPI, with a significant decrease in body weight from 2 to 3 DPI (data not shown), when mice were euthanized in extremis. Clinical signs of the neurological disease were similar between the 2 EHV-1 strains with slight differences. The signs were weight loss, ruffled hair, severe depression, head tilt, hyperexcitability, circling, ataxia, loss of proprioception, spastic paralysis of hindlimbs and tail, and recumbency. A4/72-infected mice were more depressed when compared to A9/92-infected mice, in which the other neurological signs were more prominent.
Electron Microscopy
Thin sections of OB and frontal cortex of EHV-1-infected mice were analyzed by light microscopy and the areas that exhibited histologic lesions were analyzed by EM. Ultrastructural evaluation of brain from EHV-1-infected mice revealed large numbers of cells (presumably neurons) exhibiting degenerative and necrotic changes, such as clumping and dissolution of nuclear chromatin, fragmentation of both nuclear and cytoplasmic membranes, and dissolution and vacuolation of the cytoplasm (Fig. 1). The cytoplasm of these necrotic cells usually contained numerous degenerated organelles, especially mitochondria. There was severe multifocal disruption and vacuolation of the neuropil surrounding these degenerative and necrotic cells. In less affected areas, few astrocytes and their foot processes were markedly swollen (Fig. 2). Neurons from these less affected areas exhibited normal morphology, which was characterized by numerous organelles such as mitochondria and Nissl bodies, and cytoplasmic prolongations consistent with dendrites and axons. Within these areas, the surrounding neuropil was still intact.
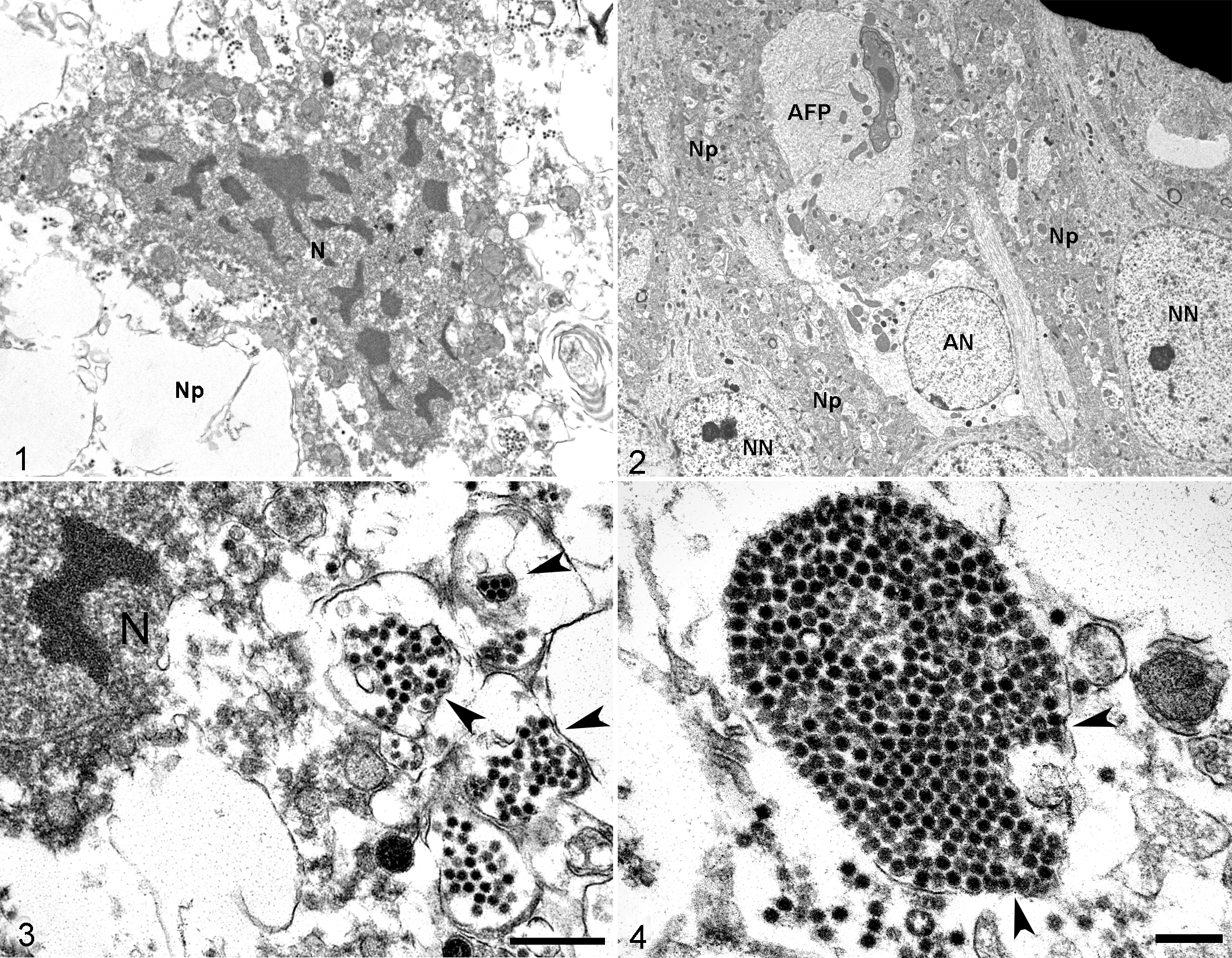
Infection with equid alphaherpesvirus 1 strains A4/72 or A9/92, brain, mouse, 3 days post inoculation. Transmission electron microscopy. N, nucleus; Np, neuropil; AN, astrocytic nucleus; AFP, astrocytic foot process; and NN, neuronal nucleus.
Viral particles were detected in sections of OB and frontal cortex of both A9/92 and A4/72-infected mice at 3 DPI. In severely affected areas of brain from EHV-1-infected mice, surrounding degenerative and necrotic cells within the vacuolated neuropil, there were numerous extracellular viral nucleocapsids, which were frequently organized in clusters (Fig. 3). Occasionally, these nucleocapsids were present within the degenerated cytoplasm, in proximity to the nucleus. The majority of viral particles were naked nucleocapsids, which frequently formed large clusters within a membrane (Figs. 4–5). Rarely, enveloped virions were detected (Fig. 6).
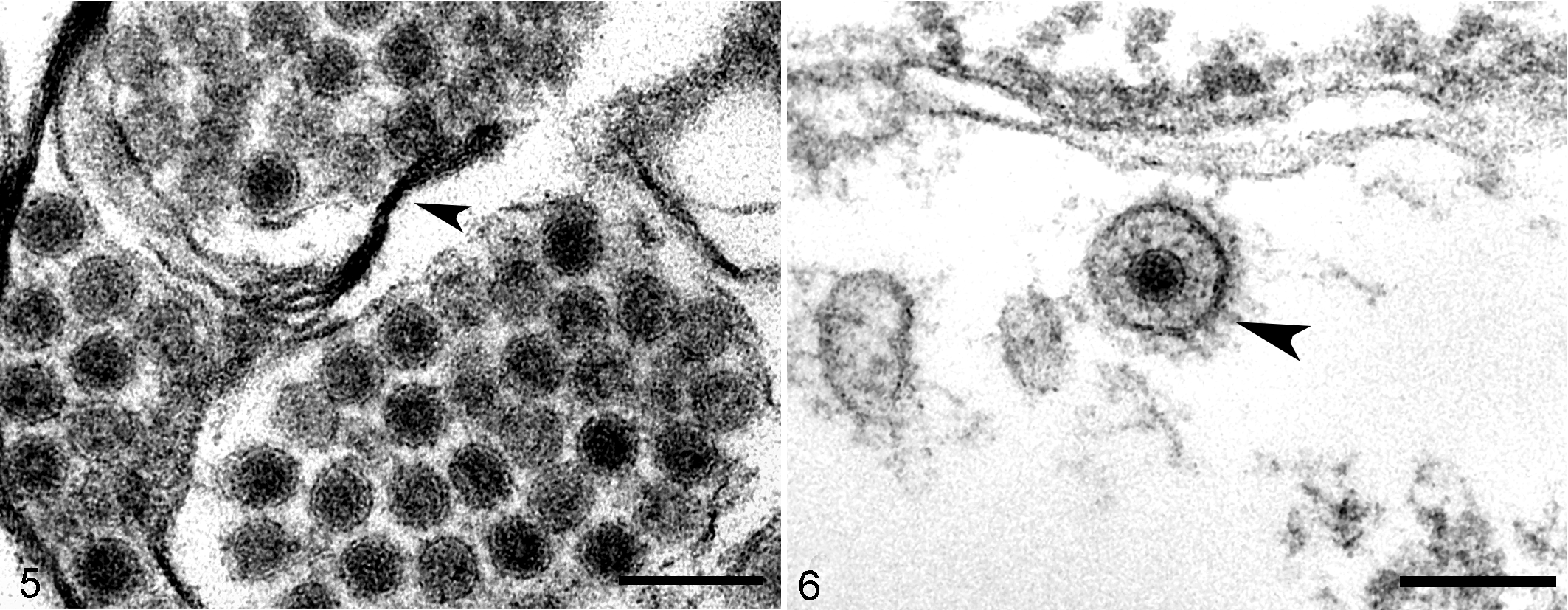
Infection with equid alphaherpesvirus 1 strains A4/72 or A9/92, brain, mouse, 3 days post inoculation. Transmission electron microscopy.
Histopathology
At 1 and 2 DPI, histologic lesions were not present within the CNS of EHV-1-infected mice. At 3 DPI, all A4/72- and A9/92-infected mice had brain lesions, which were very similar between the mice within the same experimental group. In the glomerular, outer plexiform, mitral, inner plexiform and granular layers of the OB, there was multifocal to coalescing areas with loss of neuropil and vacuolation (liquefactive necrosis; Fig. 7). In the glomerular layer, there was severe and multifocal loss of periglomerular cells (interneurons), and the remaining cells had pyknotic and karyorrhectic nuclei. There was multifocal loss of mitral cells, and the remaining cells had shrunken hypereosinophilic cytoplasm with karyorrhectic nuclei (neuronal necrosis) (Fig. 8). The granular layer was also affected, and multifocal and severe neuronal necrosis was present. Diffusely, axons extending from the olfactory nerve layer were severely disrupted, and were admixed with cellular debris and edema. In the OB of A9/92-infected mice, there were multifocal areas of hemorrhage. No lesions were present within the brain of control mice (Figs. 9, 10).
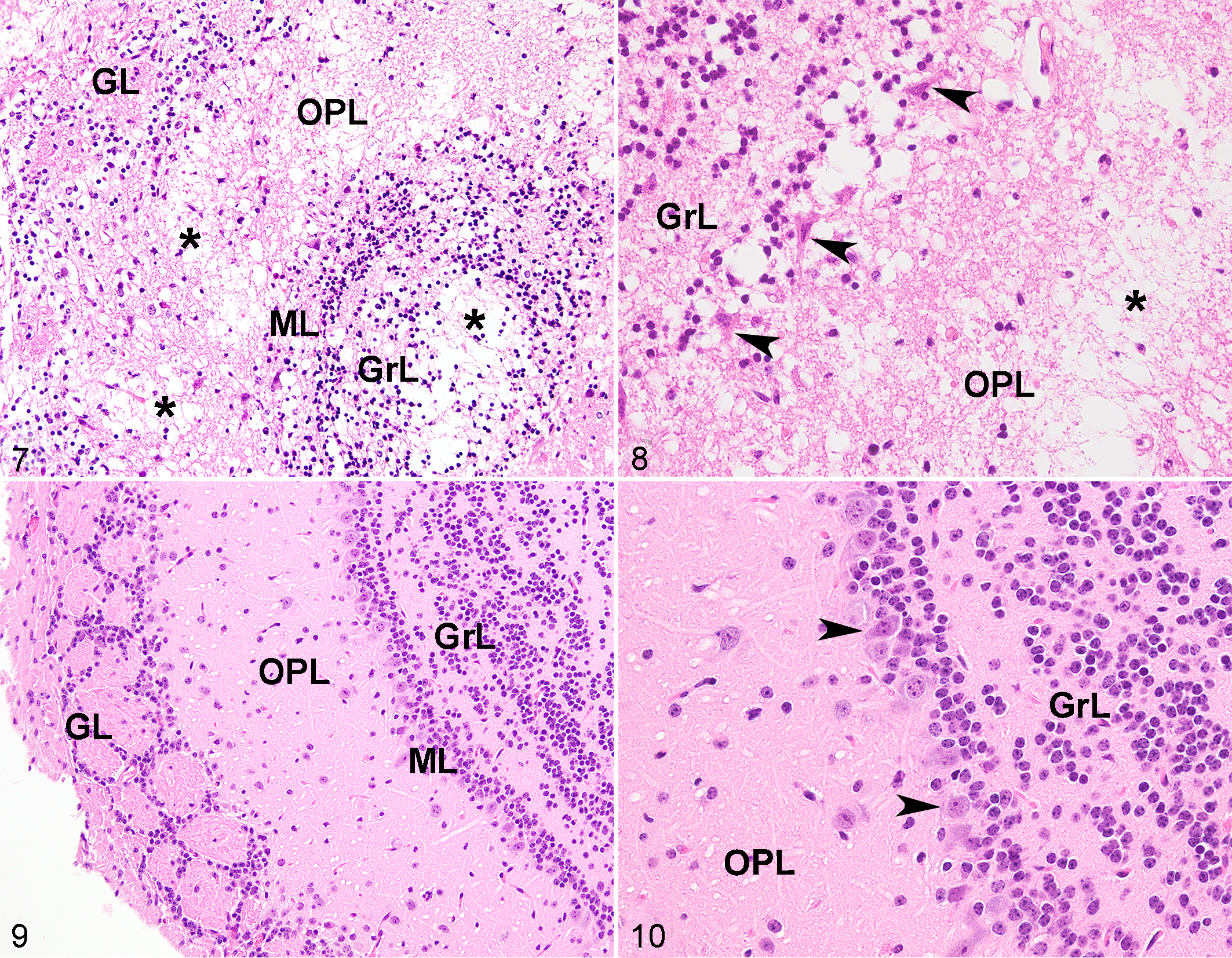
Infection with equid alphaherpesvirus 1 strains A4/72 or A9/92, brain (olfactory bulb [OB]), mouse, 3 days post inoculation. Hematoxylin and eosin (HE). GL, glomerular layer; OPL, outer plexiform layer; ML, mitral layer; GrL, granular layer.
Necrotizing and inflammatory lesions were also visualized in other areas of the CNS of EHV-1-infected mice, with differences in lesion intensity between A4/72 and A9/92 strains. Both viral strains were associated with necrotizing lesions that were similarly distributed along the ventral portion of the brain. The main affected areas were the anterior olfactory nucleus and orbital area of piriform cortex, olfactory tubercle, piriform area from striatal section to rostral mesencephalon, and entorhinal area. Within these areas, there was severe, multifocal to coalescing, neuronal degeneration and necrosis with severe pericellular edema (Figs. 11–12). There was a multifocal to coalescing loss and cavitation of the neuropil adjacent to the areas of neuronal necrosis. Neuronal necrosis with severe, multifocal to coalescing loss and cavitation of the adjacent neuropil were also visualized in other areas of the CNS of both strains, such as the caudoputamen, lateral septal nucleus, thalamus, hypothalamus and hippocampus. In control mice, lesions were not visualized within these areas (Fig. 13).
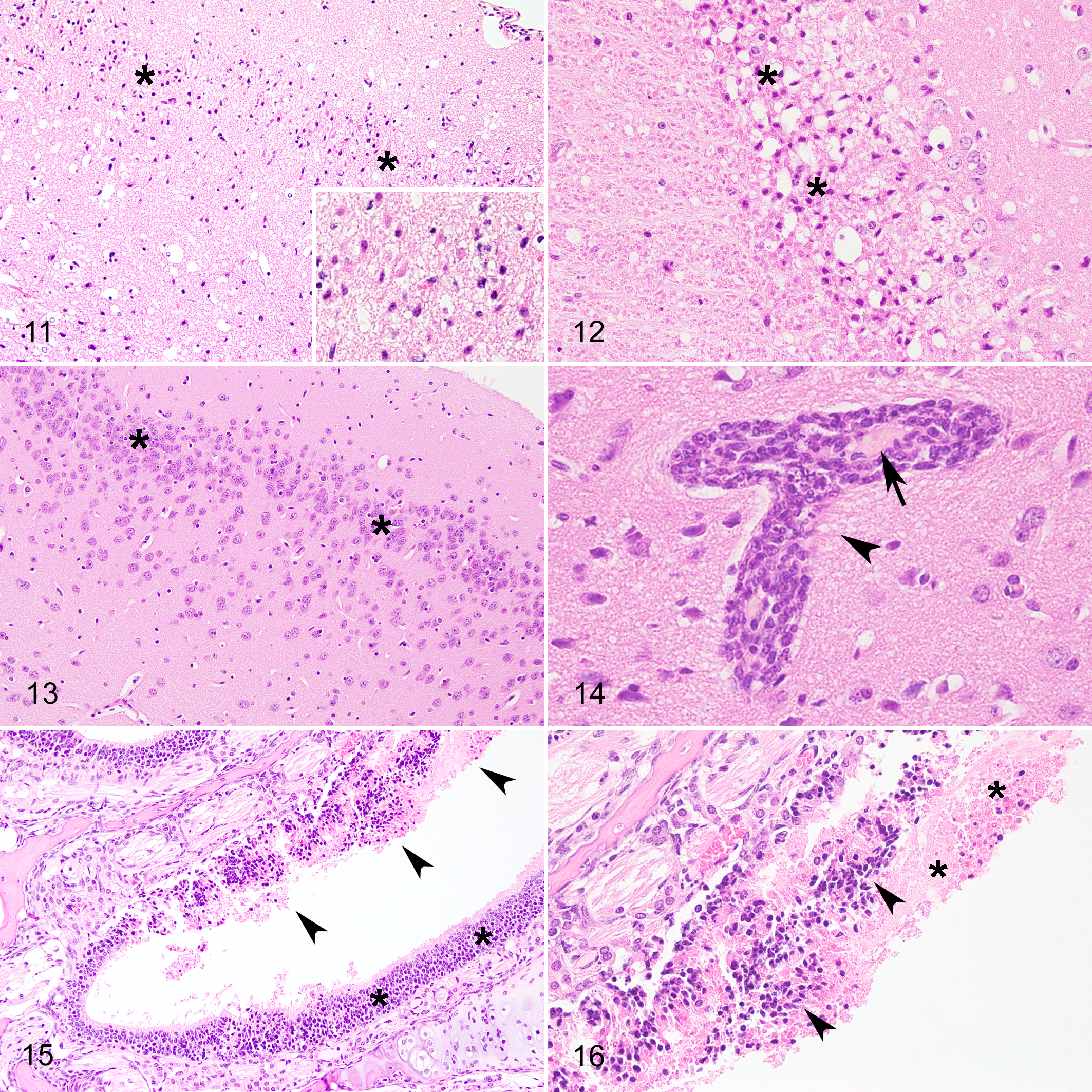
Infection with equid alphaherpesvirus 1 strains A4/72 or A9/92, brain mouse, 3 days post inoculation (DPI). Hematoxylin and eosin (HE).
In A4/72-infected mice, there was severe encephalitis in which perivascular spaces in the areas previously described were multifocally expanded by 1 to 4 layers of macrophages and lymphocytes with rare neutrophils (perivascular cuffing; Fig. 14). A large number of these inflammatory cells also expanded the leptomeninges (leptomeningitis) of A4/72-infected mice (Fig. S1). In A9/92-infected mice, perivascular cuffing was less prominent, typically a single layer of macrophages with rare neutrophils, and there was mild multifocal to coalescing leptomeningitis involving these same inflammatory cells. Overall, in the brain of both A4/72- and A9/92-infected mice, perivascular spaces were diffusely distended (edema), with hypertrophy of endothelial cells (reactive endothelium) and congestion.
In the nasal cavity of both A9/92- and A4/72-infected mice at 3 DPI, there were multifocal, locally extensive areas with marked loss of ciliated epithelium, which was replaced by large amounts of an amorphous hypereosinophilic material (necrotic cellular debris and fibrin), admixed with numerous epithelial cells that had pyknotic or often karyorrhectic nuclei (necrosis) and were frequently sloughed into the lumen of the nasal cavity (fibrinonecrotizing rhinitis; Figs. 15, 16). These necrotic areas were often associated with marked hemorrhage. Within these areas, there were rare viable and degenerated neutrophils. In EHV-1-infected mice, intranuclear inclusion bodies were not present within the olfactory epithelium or brain. No significant lesions were visualized within the lungs of EHV-1-infected mice or control mice.
Immunohistochemistry
At 3 DPI, multifocally within the brain of all EHV-1 infected-mice, perivascular spaces from neuroparenchymal and leptomeningeal blood vessels were expanded and surrounded by CD3+ cells (up to 3 layers in A4/72-infected mice or up to one layer in A9/92-infected mice), which frequently infiltrated the adjacent neuroparenchyma (Fig. 17) The CD3 immunolabeling corresponded to the areas where the perivascular cuffing was seen histologically. Moreover, CD3+ cells were identified in areas where lymphocytes were not evident histologically, showing that the inflammation was more widespread than previously described, especially in A4/72-infected mice. The areas included all those described previously, including more dorsal areas of the brain and hippocampus, with scattered clusters of CD3+ cells reaching more caudal areas of the brain such caudal mesencephalon and rhombencephalon at the level of the pons.
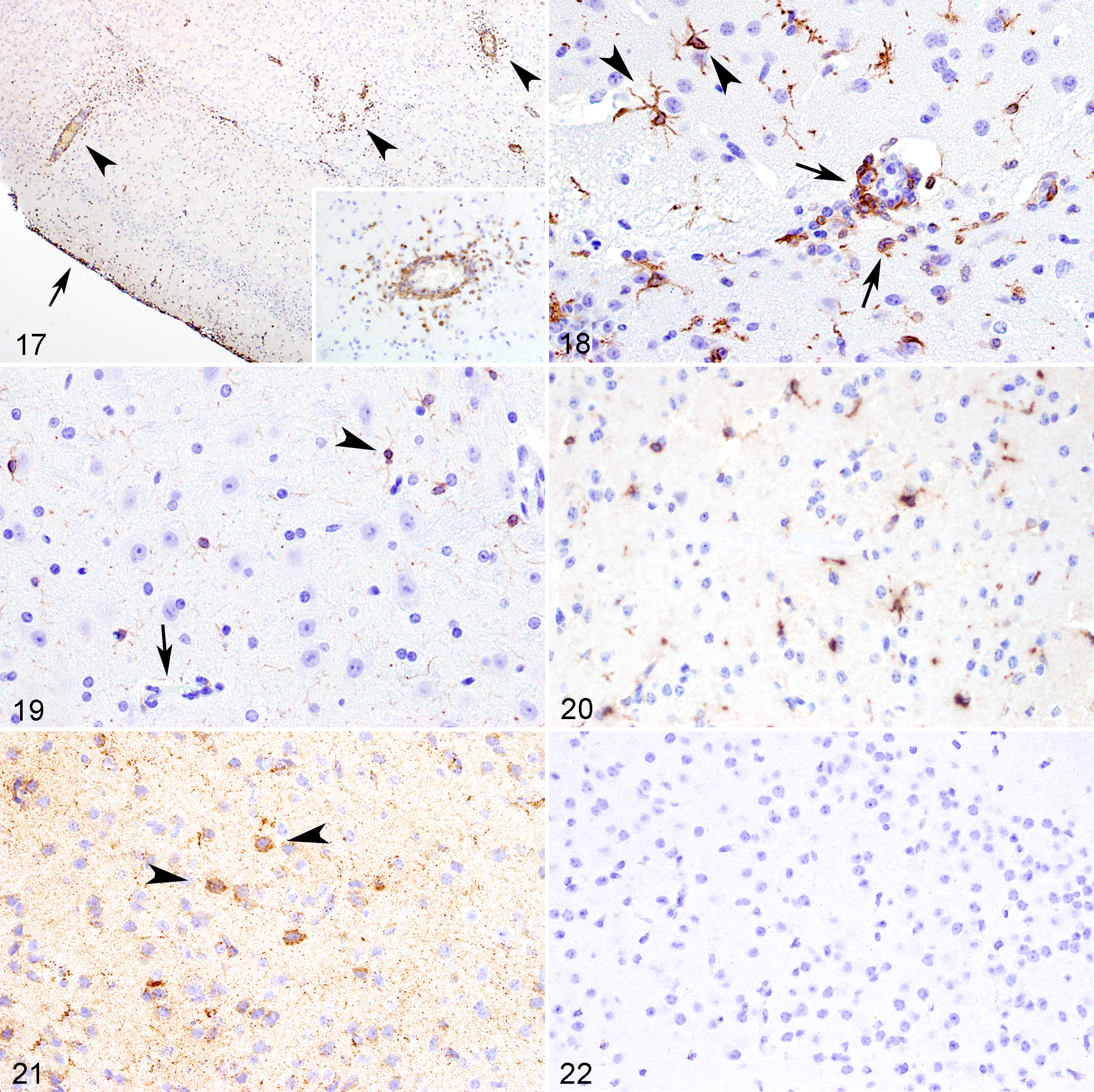
Infection with equid alphaherpesvirus 1 strains A4/72 or A9/92, brain, mouse, 3 days post inoculation.
Expression of Iba-1 (microglia and monocytes/macrophages) were evaluated in both groups of EHV-1-infected mice and control mice at 3 DPI. In all mice infected with A9/92 and A4/72 EHV-1 strains, there was diffuse immunolabeling for Iba-1 (microgliosis) in the majority of the analyzed brain areas including the OB, many areas of the cortex, septo-striatal, septo-diencephalic, caudal diencephalon (hippocampus and thalamus), mesencephalon (caudal hippocampus and rostral colliculus), and rhombencephalon (pons and medulla; Fig. 18). These areas were characterized by an increased number of Iba-1 immunolabeled cells exhibiting a morphology of activated microglia, where the microglial processes were thickened and the nuclei were enlarged and rounded. 19,22
In addition to microgliosis, in some areas of the brain from EHV-1-infected mice, the perivascular spaces of neuroparenchymal and leptomeningeal vessels, and occasionally the surrounding neuroparenchyma, contained low to moderate numbers of round cells without cellular processes that were immunolabeled for Iba-1 (Fig. 18), which were morphologically distinct from activated microglia. 24
Control mice had normal, nonactivated microglial morphology (ramified microglia), characterized by a nucleus with a more fusiform shape, and thin, ramified cellular processes (Fig. 19).
The chemokines CCL5 and CCL2 were both detected by IHC in the brain of all mice infected with A4/72 and A9/92 EHV-1 strains at 3 DPI, as homogenous immunolabeling within the cytoplasm of cells either morphologically consistent with neurons or glial cells, or within the cytoplasm of mononuclear cells within the perivascular cuffs (Fig. 20). Immunolabeling for CCL5 (Suppl. Figs. S2–S7) and CCL2 (Suppl. Fig. S8–S11) was widely distributed within the brain of EHV-1-infected mice, and both chemokines had a similar distribution. Immunolabeling was more intense in areas where there were significant histologic lesions. However, CCL5 and CCL2 were also detected in many areas where histologic lesions were not present. These areas included all layers of the OB, and many areas of the cortex, basal nuclei area, lateral septal nucleus, thalamus, hypothalamus, hippocampus, caudal mesencephalon and rhombencephalon at the level of pons.
Immunolabeling for cleaved caspase-3 was evaluated in both EHV-1-infected mice and control mice at 3 DPI, and the patterns were similar in mice infected with the A4/72 and A9/92 EHV-1 strains. Caspase-3 immunolabeling was present in areas where necrotizing lesions were detected, as well as other areas with mild histologic lesions. Immunolabeling was present in the OB, many areas of cortex including the frontal cortex up to the occipital lobes, basal nuclei area, thalamus and hypothalamus, hippocampus, and caudal mesencephalon. In areas of the brain with necrosis and degeneration, caspase-3 immunolabeling was granular and mainly in the neuroparenchyma adjacent to necrotic neurons. In the areas where significant histologic lesions were not present, caspase-3 immunolabeling was detected as strong, granular, cytoplasmic staining mainly in neurons (Fig. 21), and in cells with glial morphology. No strong caspase-3 immunolabeling was detected in control mice (Fig. 22).
Discussion
Intranasal inoculation of A4/72 and A9/92 EHV-1 strains induced neurological disease in C57BL/6J mice that was invariably fatal by 3 DPI, and was characterized by severe necrotizing, lymphocytic and histiocytic encephalitis with extensive neuronal necrosis, microgliosis, and expression of CCL2, CCL5, and caspase-3. By electron microscopy, there were numerous viral nucleocapsids and fewer enveloped virions within degenerated and necrotic neurons and in the surrounding neuropil. The lesions were mainly detected in the OB and extending along the ventral portions of the brain and meninges. Although EHV-1 affects mainly the respiratory system of mice, 4,5,26,51 and eventually demonstrates a certain level of neurotropism, 4,5,15,18,41 A4/72 and A9/92 EHV-1 exhibited high neurovirulence in mice.
Resident cells of the brain can recognize and initiate an innate immune response after infection by neurotropic viruses. 38 Cytokines and chemokines, secreted by either resident cells of the brain or infiltrating leukocytes, can recruit inflammatory cells that cross the blood-brain barrier and infiltrate the brain causing encephalitis. 6 In the present study, in the areas where the virus was detected, there were extensive areas of neuronal necrosis as well as perivascular cuffing of CD3 positive T cells and Iba-1 positive cells with morphology most consistent with monocytes. 24 Concomitantly, within these areas, there was significant expression of CCL2 and CCL5 chemokines in both neurons and glial cells. These chemokines, among others, are considered critical regulators of leukocyte trafficking into the CNS during viral infections. 21 CCL-2 and CCL-5 are mainly involved in the infiltration of monocytes and T lymphocytes into the neuroparenchyma. 43,44 In addition, CCL2 is associated with infiltration of NK cells to the site of infection. 10 CCL2 and CCL5, as well as other chemokines, were also expressed in the lungs of EHV-1-infected mice, 26,48,51 as well as in equine airway epithelial cells 49,55 and equine endothelial cells 25 infected by EHV-1 in vitro.
Although the expression of chemokines and cytokines with subsequent infiltration of inflammatory cells into the CNS might act to control viral replication in some situations, the immune response against viruses could also result in CNS damage. 27,50 In the present study, there were numerous reactive microglial cells in the areas of inflammation and necrosis. Microglia have antiviral effects such as phagocytosis and secretion of cytokines, 8 but microglial activation can induce damage to the CNS. 8,31 In addition, infiltrating inflammatory cells are correlated with increased CNS pathology and mortality rates in humans alphaherpesvirus 1, 30 West Nile virus, 14 and Theiler’s murine encephalomyelitis virus infections. 7 The results of the current study suggest that pro-inflammatory chemokines along with productively EHV-1 replication in neurons might contribute to the extensive necrotic and inflammatory lesions observed in EHV-1-infected mice. Moreover, numerous neurons were immunolabeled for activated caspase-3, suggesting that in addition to necrosis, EHV-1 might trigger neuronal death by apoptosis.
The mouse model of EHV-1 infection presented here differs from others, since A4/72 and A9/92 EHV-1 strains exhibited a higher neurotropism with 100% mortality. 4,5,18 Comparatively, Ab4, which is another EHV-1 neurotropic strain for mice, induced 50% mortality at 3 DPI at a high inoculation dose. 15 A4/72 and A9/92 strains also induced severe meningoencephalitis in other mouse strains, 36 as well in hamsters, 33 with similar lesions in the CNS. Moreover, in the present study, A4/72 and A9/92 EHV-1 strains did not induce any inflammatory or necrotizing lesions in the lower respiratory tract of mice. This lesion pattern differs from other strains of EHV-1 which do induce pulmonary lesions in mice. 4,5,13,26,48,51 The lack of lower respiratory lesions in A4/72 and A9/92 suggests that these viral strains have neuropathogenicity that may be explained by differences in the viral genome. For instance, in horses, a single polymorphism in nucleotide 2254 of DNA polymerase gene (ORF30) was associated with neurological disease. 39 However, the A4/72 and A9/92 strains were previously classified as non-neuropathogenic based on genotype (A2254; amino acid N752), 33 demonstrating that other genes and factors might play a role in neuropathogenicity in mice.
The distribution of histologic and ultrastructural lesions in A4/72 and A9/92-infected mice suggested that EHV-1 induced a necrotizing rhinitis, reaching the brain through the olfactory pathway, and infecting mainly neurons in the CNS, as described by others. 15 In contrast, in naturally infected horses, EHV-1 initially infects epithelial cells within the nasal mucosa, later infecting lymphocytes and monocytes in the regional lymph nodes. 29 With cell-associated viremia, the infected leukocytes, once in brain capillaries, carry the virus to endothelial cells and stimulate an immune response that results in thrombosis, inflammation and necrosis. 42 Previous studies demonstrated that mouse endothelial cells, differently from equine endothelial cells, are not susceptible to EHV-1 infection. 17,18 Therefore, in the mouse model presented here, EHV-1 affects primarily neurons, resulting in neuronal necrosis and inflammation, which significantly differs from the natural disease in horses.
Although EHV-1 infect mainly equids, the virus has the potential to infect other animal species. 54 EHV-1 (or EHV-9, which is closely related to EHV-1) has been implicated as the cause of fatal neurological disease in non-equid species from zoos including endangered species such as in Indian rhinoceros, polar bears, black bears, Thomson’s gazelles, and guinea pigs. 1,11,16,45,46,53 The lesions in these non-equid species are generally characterized as an encephalitis or meningoencephalitis, with neuronal necrosis and neuronal infection, and without evidence of endothelial tropism. 11,45,46,53 Thus, the lesions caused by EHV-1 or EHV-9 in non-equid species are very similar to A4/72 and A9/92 EHV-1 infection in mice, and similar changes were also described in the hamster model. 12,33 Therefore, the model presented here might also be useful in aiding to understand the pathogenesis of EHV-1 infection in non-equid species.
Supplemental Material
Supplemental Material, sj-pdf-1-vet-10.1177_03009858211020670 - Pathogenesis of Equid Alphaherpesvirus 1 Infection in the Central Nervous System of Mice
Supplemental Material, sj-pdf-1-vet-10.1177_03009858211020670 for Pathogenesis of Equid Alphaherpesvirus 1 Infection in the Central Nervous System of Mice by Leonardo P. Mesquita, Rafael C. Costa, Laís L. R. Mesquita, Maria do Carmo C. S. H. Lara, Eliana M. C. Villalobos, Claudia M. C. Mori, Enio Mori, Elizabeth W. Howerth and Paulo C. Maiorka in Veterinary Pathology
Footnotes
Acknowledgements
The authors would like to acknowledge Mary Ard for her excellent technical assistance with ultrastructural analysis, and we are also thankful to the staff from the Histology Laboratory from Department of Pathology, UGA, for their outstanding help with immunohistochemistry.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was funded by Sao Paulo Research Foundation (FAPESP), Process Nos. 2016/09396-2, 2016/24856-0, and 2018/23619-0.
Supplemental Material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.