
Abstract
A neurological disease was investigated in 3 German Shepherd pups from the same litter that failed to grow normally, appeared stiff, were reluctant to move, and were deaf. They developed intermittent seizures and ataxia and had proprioceptive defects. Histopathology showed severe vacuolation of neurons, astrocytes in nervous tissue, renal tubular epithelial cells, and macrophages in nervous tissue, spleen, and liver. Vacuoles appeared empty with no storage material stained by periodic acid–Schiff (PAS) or Sudan black stains, leading to a diagnosis of a lysosomal storage disease and in particular an oligosaccharidosis. Biochemical and genomic studies showed that this was β-mannosidosis, not previously diagnosed in dogs. A c.560T>A transition in exon 4 of the MANBA gene was found, which segregated in these and other family members in a manner consistent with it being the causative mutation of an autosomal recessive disease. This mutation led to substitution of isoleucine to asparagine at position 187 of the 885 amino acid enzyme, a change expected to have functional significance.
Keywords
There are more than 40 lysosomal storage diseases (LSDs) of humans, many of which are also found in domestic animals. 3,17,20 Most are caused by a primary lysosomal hydrolase defect, but others may be associated with posttranslational processing defects of lysosomal enzymes, defects in trafficking of lysosomal enzymes, defects in lysosomal enzyme protection, defects in other associated nonenzyme proteins (eg, activator proteins), or defects in nonenzymatic transmembrane proteins. 20 Most lysosomal storage diseases are inherited as autosomal recessive traits, but 2 sex-linked disorders, mucopolysaccharidosis II and α-galactosidosis (Fabry disease), occur in human patients. 3,17 There are also a small number of acquired diseases mainly found in domestic animals, associated with toxic substances in plants or pharmaceutical products that interfere with lysosomal function. 3
Deficient activity of a lysosomal hydrolase leads to enlargement of the lysosomal compartment by accumulation of the enzyme’s substrate(s). Far from being a passive event, this invariably sets off a cascade of secondary changes resulting in manifestations of disease. 21,25 Although usually generalized, most lysosomal diseases affect the central nervous system, and presenting signs are typically neurological, sometimes including blindness secondary to retinal degeneration. Other important signs in certain diseases that may assist diagnosis are abnormal bone development, hepatomegaly, corneal opacity, or inclusions in lymphocytes or neutrophils. 3,17,20
The histopathologic and electron microscopic appearance of storage cytosomes is only occasionally specific for a disease but is usually highly informative despite overlap occurring between various classes of disease. The biggest group are the lipidoses in which cytosomes tend to be sudanophilic with ultrastructural findings of electron-dense granular and membranous profiles. These are mostly distinct from, but somewhat similar to, those of the neuronal ceroid-lipofuscinoses. The glycoproteinoses (oligosaccharidoses) are mostly characterized by membrane-bound empty vesicles as their water-soluble contents are leached during fixation and preparation of tissue sections for microscopy. The mucopolysaccharidoses and mucolipidoses may share both types of storage lesion. The diagnosis of a lysosomal disease may be narrowed by assessing the clinical nature of the disease and the gross and microscopic appearance of lesions. Specific diagnosis depends on biochemical and molecular genetic investigation. 8,26
This report describes β-mannosidosis in German Shepherd dogs. This is a very rare inherited lysosomal disease of humans and otherwise only reported in Nubian goats 9,10 and Salers cattle. 1,4,7 A knockout mouse model has also been described. 28
Materials and Methods
Microscopy
Tissues (from case Nos. 1 and 2) were fixed in 10% buffered formalin and embedded in paraffin, and sections were stained with hematoxylin and eosin (HE), periodic acid–Schiff (PAS), Sudan black, and Luxol fast blue methods. Formalin-fixed brain, liver, and kidney from case No. 1 were also embedded in epoxy resin and thick sections stained with Toluidine blue.
Thin-Layer Chromatography
Thin-layer chromatography was performed on silica gel and stained with orcinol according to the method of Sewell. 22
Enzymology
Dog liver (control Huntaway dog and case No. 1 sample) was homogenized in 0.5 M NaCl/0.02 M Tris buffer (pH 7.4). Total protein content/mg tissue was determined using a Pierce BCA Kit (#23225; Thermo Fisher Scientific, Waltham, MA). The activity of 3 lysosomal enzymes (α-D-mannosidase [EC 3.2.1.24], β–D-mannosidase [EC 3.2.1.26], and α-L-fucosidase [EC 3.2.1.51]) was measured using the fluorogenic substrates, 4-methylumbelliferyl (MU)–β-D-mannopyranoside (#N2127; Sigma-Aldrich, Merck KGaA, Darmstadt, Germany), 4-MU-β-D-mannopyrannoside (#M0905; Sigma-Aldrich), and 4-MU-α-L-fucopyranoside (#8527; Sigma-Aldrich), respectively. Substrate was added to tissue homogenate (10 μg protein equivalent) and incubated at 37°C for 30 minutes at either pH 4.5 for α-mannosidase or pH 5.0 for β-mannosidase and α-fucosidase. The reaction was stopped with 0.1 M glycine (pH >10.8) and the liberated 4-MU measured using a luminescence spectrometer (Victor 3 multiplate reader, #1420; Perkin Elmer, Waltham, MA, USA). Activity is reported in pmol/min/mg total protein.
Molecular Genetics
DNA to be used for whole-genome sequencing was extracted from the stored liver of 1 affected dog (case No. 1) and the stored heparinized blood of the dam, using the MagAttract HMW genomic DNA extraction kit (Qiagen GmbH, Hilden, Germany) as per the manufacturer’s instructions. DNA to be used for genotyping was extracted from stored liver of the second affected dog (case No. 2) and from stored blood of the sire, 1 unaffected sibling, 2 dogs from same breeder but different parentage, and 1 normal unrelated German Shepherd dog using the DNeasy Blood and tissue kit (Qiagen GmbH) as per the manufacturer’s instructions.
The high-quality, high-molecular-weight DNA was sent to the Iowa State University DNA sequencing facility (Ames, IA), where a fragment library was prepared using the Illumina TruSeq DNA PCR free library preparation kit (Illumina, San Diego, CA) with a 550-bp insert size. Two lanes of paired-end reads (2 × 100 bp) were then obtained using an Illumina HiSeq 3000 machine (Illumina).
The fastq files were aligned to the reference dog genome (CanFam3.1) using BWA-MEM 0.7.15. 12 The sequences have been uploaded to the NCBI sequence read archive (SRA) with SRA accession SRP159211, project number PRJNA486428, and sample numbers SAMN09845873 (case No. 1) and SAMN09845874 (Dam).
Variants were called using GATK 3.4–46 HaplotypeCaller. 15 Joint genotyping was performed by merging the individual g.vcf files for each animal using GATK GenotypeGVCFs mode with the default parameters. Percentage of reads mapped was determined using Samtools flagstat. 13 Read coverage was determined using GATK DepthOfCoverage mode.
Genetic variant annotation and effect were predicted using Ensembl Variant Effect Predictor (VEP 90.3) and reference genome CanFam3.1 with --sift prediction. 16 The results were then filtered using VEP 90.3 filter mode for all genes associated with lysosomal storage diseases and variants within these genes considered deleterious by SIFT. 16
The putative mutant variant was genotyped by positional sequencing using polymerase chain reaction (PCR) followed by Sanger sequencing. Primers were designed using Geneious 8.1 (Biomatters Ltd., Auckland, NZ) and the Primer 3 algorithm to span the variant, with a forward primer sequence of 5′-TTGAGCTGCGTTTCCAGTCA-3′ and a reverse primer sequence of 5′-CATCCCATGAAGCAGTCAGGA-3′, resulting in a 260-bp fragment. The PCR mix contained 1× PCR buffer, 1.5 mM MgCl2, 0.2 mM each dNTP (Fisher BioTec Ltd., Wembley, WA, Australia), 0.2 mM each primer, 1 unit Taq-Ti DNA polymerase (Fisher BioTec Ltd.), and 2 μL DNA, made up to 25 μL with H2O. The PCR conditions were as follows: 95°C for 10 minutes followed by 40 cycles of 95°C for 30 seconds, 60°C for 30 seconds, and 72°C for 30 seconds, with a final extension of 72°C for 5 minutes and then chilled at 4°C using an Applied Biosystems Veriti Thermal Cycler (Applied Biosystems, Foster City, CA). The purified amplicon with the PCR primers was sent to the Massey University Genome Service (Massey University, Palmerston North, New Zealand) and subjected to an automatic dye-terminator cycle sequencing with BigDye Terminator version 3.1 Ready reaction cycle sequencing kit and the ABI3730 Genetic Analyzer (Applied Biosystems). The sequence data were analyzed using Geneious 8.1.
Results
History and Clinical Presentation
A male German Shepherd pup (case No. 1) was obtained from a breeder at 8 weeks of age. Over subsequent weeks, the new owners noticed that he grew slowly, failed to lose deciduous teeth, and appeared deaf. He walked but never ran and required assistance to overcome obstacles. Case No. 1 was euthanized at 8 months of age, at which point on physical examination he was stiff with extensor weakness in neck and hind limbs and had hind limb ataxia and conscious proprioceptive deficits in all 4 limbs. There were intermittent seizures. He had a depressed mental attitude, appeared to be in pain, and was reluctant to move. Two further pups from the same litter of 11 pups were found to have similar clinical signs and were also euthanized at 8 months of age. Fixed tissue was submitted for further analysis from one of the additional affected pups (case No. 2).
Histopathology
Histological changes in formalin-fixed, HE-stained sections showed severe foamy vacuolation of neurons (Fig. 1). In many large neurons in the brainstem and Purkinje cells in the cerebellum, vacuoles appeared relatively discrete and up to 1 to 2 μm in diameter. In the cerebrum, vacuolation was so severe that cortical neurons appeared ballooned with few cytoplasmic elements surviving the paraffin-embedding process. In epoxy resin sections stained with Toluidine blue, some cells had a myriad of small vacuoles giving the cell a foamy appearance, whereas in others, the vacuoles were larger and more discrete (Figs. 2, 3).
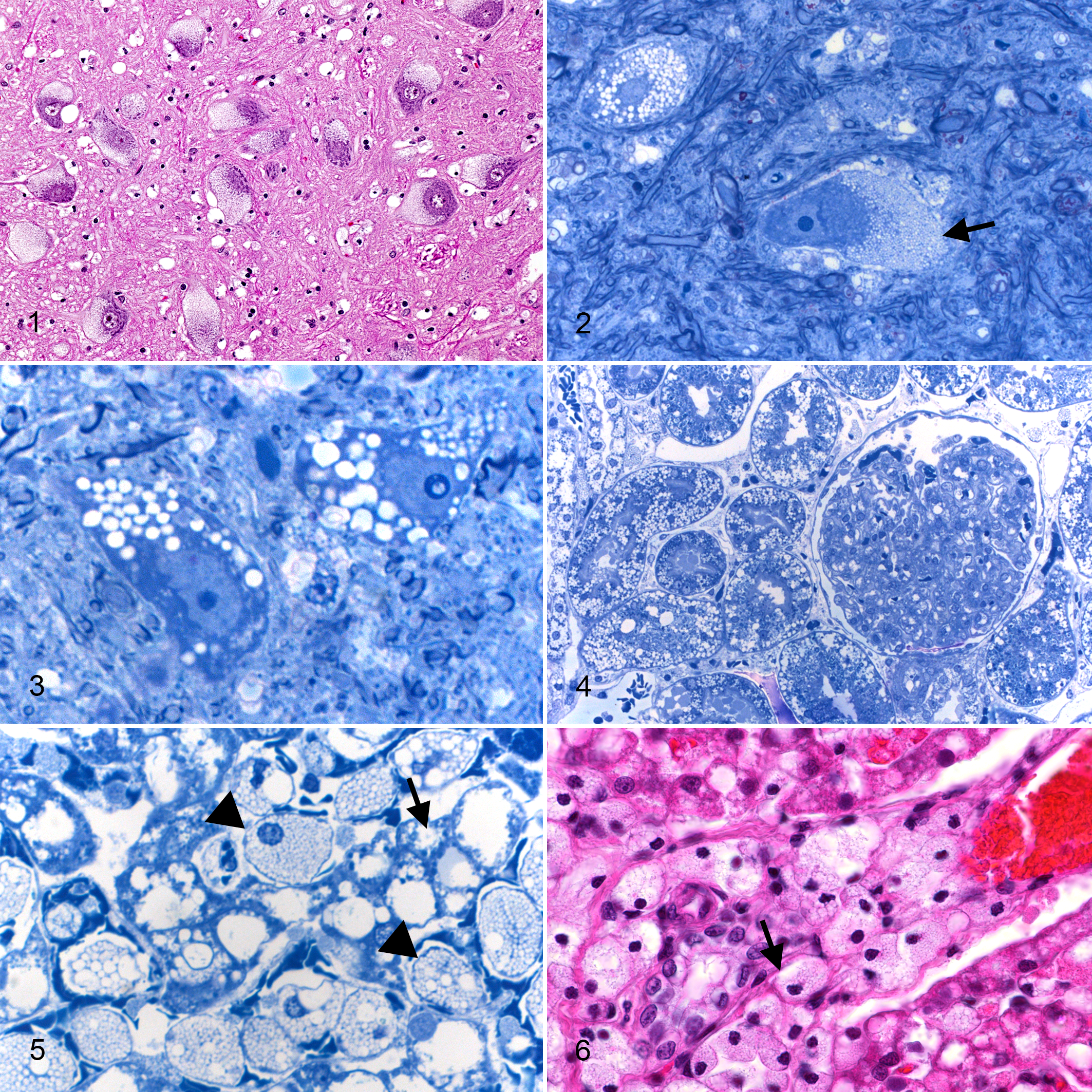
Cells interpreted as astrocytes were swollen, and there were many vacuolated macrophage-like cells as neuronal satellite cells (Fig. 2) in perivascular spaces and in the meninges. Storage material did not stain with PAS, Sudan black, or Luxol blue stains. Luxol blue staining showed normal myelination.
Nerves in the brachial plexus were heavily infiltrated by macrophages, many of them highly vacuolated. The layers of the retina were intact, but there was swelling of Müller cells and vacuolation of ganglion cells. The cornea was normal.
Severe vacuolation of macrophages was present in lymph nodes and spleen. In the kidney, most parts of the nephron unit were vacuolated, more clearly seen in epoxy resin sections (Fig. 4). In the liver, there was some vacuolation of hepatocytes and Kupffer cells, but the dominant change was the presence of many large globular vacuolated macrophages in the sinusoids (Fig. 5), around central veins and within the portal areas (Fig. 6). Many heavily vacuolated macrophages occurred in the peribronchial lymphatics of the lung.
Thin-Layer Chromatography
Analysis of urinary oligosaccharides yielded a strongly staining and highly mobile band in the affected dog urine (case No. 1) that was not in the urine of a control dog. Two other highly mobile bands occurred in both control and affected dog urine. In addition, several less staining bands were also seen, some of which only occurred in the urine of the affected dog (Fig. 7).

Orcinol-stained thin-layer chromatograph of urine from an affected and a normal control dog shows a large strongly staining band (arrowhead) running just behind 2 normal bands. Faintly staining but distinct slower-running bands are also shown in the affected urine, not all appearing in the control. The thin-layer chromatography of urine was interpreted as showing excretion of an abnormal low-molecular-weight oligosaccharide, supporting the histological evidence of an oligosaccharidosis.
Enzyme Assays
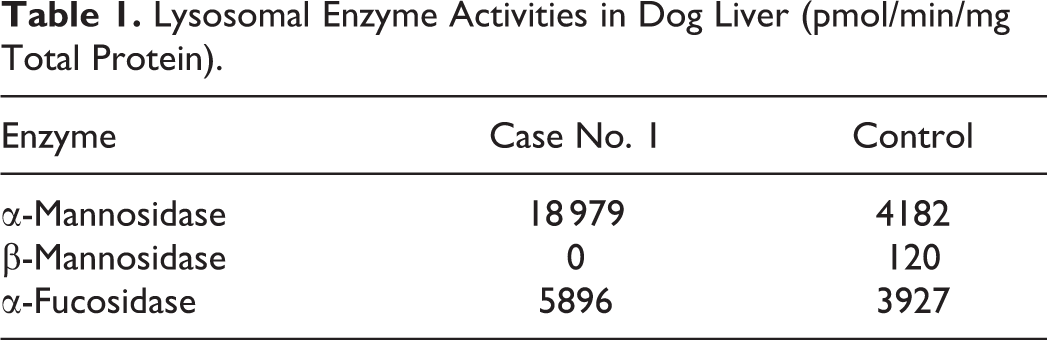
The enzyme activities for 3 lysosomal enzymes are shown in Table 1. There was lack of β-mannosidase activity compared to the normal control dog.
Lysosomal Enzyme Activities in Dog Liver (pmol/min/mg Total Protein).
Whole-Genome Sequencing and PCR Testing of Family
Whole-genome sequencing of 2 affected dogs resulted in 2.4 to 2.5 Gbp of sequence data, as summarized in Supplemental Table S1. Filtering the variants that were in genes associated with lysosomal storage disease, considered to be deleterious using SIFT, and were heterozygous in the dam of case No. 1 and homozygous mutant in case No. 1 resulted in 1 variant, a nonsynonymous (missense) variant in the MANBA gene.
The variant was a T>A transition at CFA32:24147500 (CanFam3.1) in exon 4 of the MANBA gene. The c.560T>A variant resulted in a change of amino acid from an isoleucine to an asparagine at position 187 of the 885 amino acid β-mannosidase (MANBA) protein (p.I187 N). This missense mutation was not a known variant in the Ensembl Canis familiaris database (www.ensembl.org/Canis_familiaris) or the European Variant Archive (www.ebi.ac.uk/eva). The mutation occurs in a highly conserved area of the β-mannosidase protein (Fig. 8), which is predicted to form a beta bridge (Phyre, RCSB.org) in chain B of the LacZ (part of the glycosyl hydrolase family 2, COG3250, PDB ID 2JE8) domain.
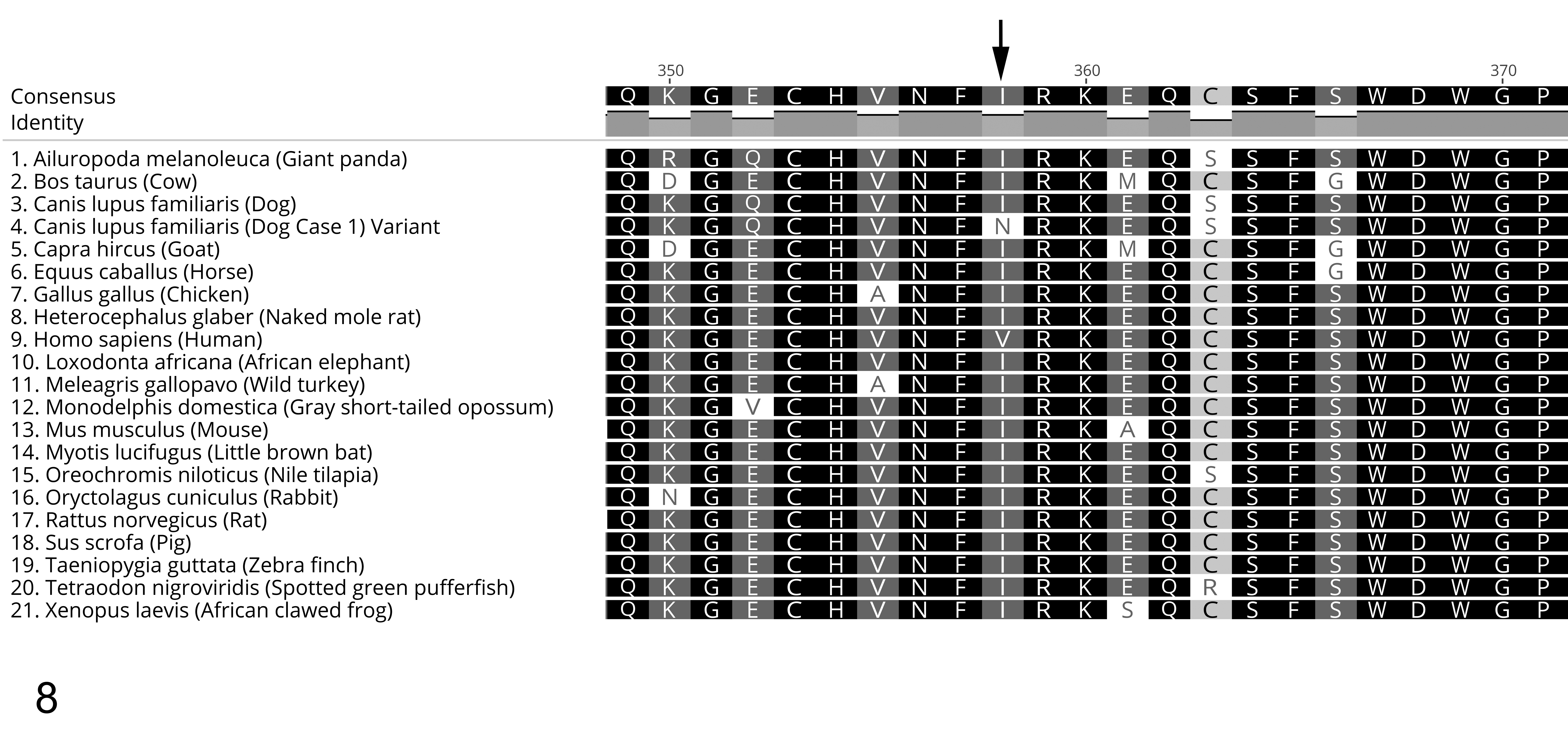
Alignment of MANBA protein sequences from 20 species, plus the p.I187 N variant (arrow) in affected German Shepherd dogs (case No. 4). Note the conserved nature of this part of the protein, as indicated by the nearly identical amino acid sequence in the different species.
The variant was validated using PCR and Sanger sequencing of DNA from related and unrelated dogs. The 2 parents and 1 littermate were heterozygous for the mutation, the 2 affected dogs (case Nos. 1 and 2) were homozygous for the mutation, and 2 other dogs from the same breeder but of different parentage were homozygous wild-type (Suppl. Fig. S1). An unrelated German Shepherd dog was also homozygous wild-type.
Discussion
A lysosomal storage disease was diagnosed based on histopathology and, in particular, an oligosaccharidosis, as storage material appeared to be soluble and lost during tissue processing and the histopathology was most similar to other oligosaccharidosis, rather than the mucopolysaccharidoses or mucolipidoses. 8,26 There was no reason to expect other than a genetic etiology for this disease; 3 affected dogs out of a litter of 11 (27%) is consistent with a recessive mode of inheritance if both parents are carriers. The gross and histological features did not fit well with those of mucopolysaccharidosis VII (MPS VII), a known lysosomal storage disease of German Shepherd dogs. 23 Animals affected with MPS VII typically show corneal opacity, dwarfism, and extreme joint laxity, none of which were present in the dogs in the present study. 6,23 Animals with MPS also have secondary storage of lipids and gangliosides that undergo fluorescence; this was absent in these German Shepherd dogs. 14
Oligosaccharidoses result from defective catabolism of complex, hybrid, and high-mannose oligosaccharides that are N-linked to proteins through asparagine (Asp) to form glycoproteins. These glycans have a common core structure of Asp, N-acetylglusoamine (GlcNAc), fucose (Fuc), and mannose (Man) (Fig. 9). This is normally α-linked to 2 mannose units, which are linked in turn to a variety of other sugars in a branched formation.
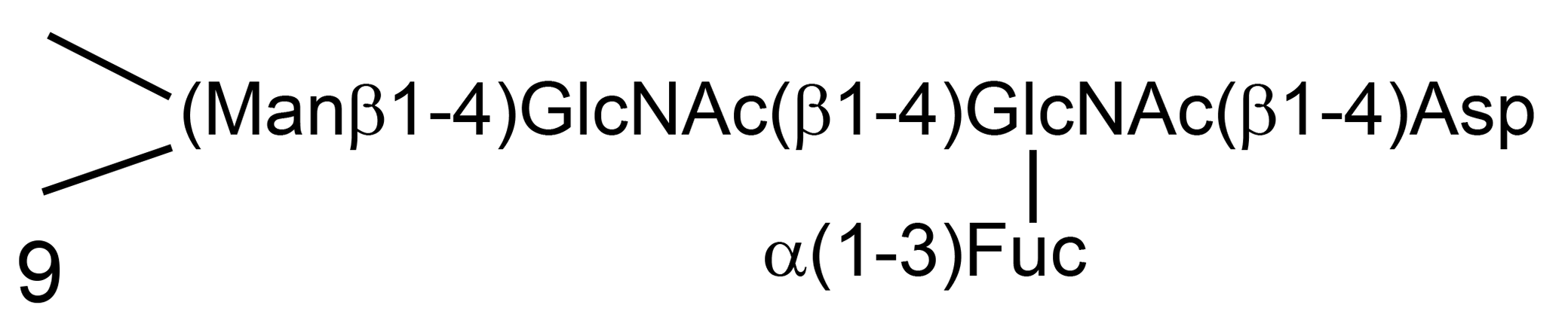
The structure of the core region of N-linked oligosaccharides of glycoproteins. 27
Lysosomal catabolism of the oligosaccharides occurs bidirectionally in which the first step is the cleavage of asparagine from the protein. The second step is the cleavage of asparagine and then fucose from the core and the branched chains. Only then does catabolism occur in an ordered sequential manner from the other nonreducing end of the larger oligosaccharide. 27
Thin-layer chromatography of urine was interpreted as showing excretion of an abnormal low-molecular-weight oligosaccharide running just behind the first 2 normal bands, supporting the histological evidence of an oligosaccharidosis. Several other distinct but lesser stained bands are also shown, only some of which appear in the control urine (Fig. 7). Because of the enlarged spinal nerves, not described in other canine LSDs, fucosidosis was an initial provisional diagnosis. 5,11 β-Mannosidosis was also considered a likely diagnosis based on the excretion of major low-molecular-weight oligosaccharides, Man(β1–4)GlcNAc(β1–4)GlcNA and Man(β1–4)GlcNAc(β1–4)GlcNAc(β1–4)GlcNAc, in both affected goats and Salers cattle. 1,9 In other oligosaccharidoses including fucosidosis, bands of different running rates would be expected, 2,18 so these diseases were thought less likely prospects.
A diagnosis of β-mannosidosis was established by enzymology and confirmed by a parallel whole-genome sequencing investigation in which the mutation c.560T>A was found in a highly conserved region of the MANBA gene. Isoleucine is a nonpolar, uncharged aliphatic amino acid, while asparagine is a polar amino acid, and multiple amino acid substitution prediction algorithms suggested the mutation would affect protein function, as confirmed by enzymology. The segregation pattern of the mutation in affected animals (Fig. 9), parents, and other offspring supported the conclusion that this was the causative mutation. This is the first diagnosis of β-mannosidosis in dogs and the fourth species in which it has been shown to occur naturally.
There is considerable heterogeneity in age of onset and clinical course in human patients, some of whom live into their late middle age. They have mental retardation, joint stiffness, and deafness, and they may have minor skeletal abnormalities, including fascial dysmorphism and hepatosplenomegaly (OMIM 248510). In Nubian goats and Salers cattle, the disease is neonatal with affected individuals unable to rise and walk. In goats, there is a dome-shaped skull, small palpebral fissures, carpal contractures, hyperextension of the pastern joints, ocular oscillations, intention tremor, and deafness. Histologically, in addition to vacuolation of neurons and cells in a wide variety of other tissues, there is hypomyelination of white matter. 9,10 The disease is similar in Salers calves, including hypomyelination but also enlarged kidneys. 1,4,19 Hypomyelination was not a feature of the present canine case.
β-Mannosidosis of German Shepherd dogs more closely resembles the clinical pattern in humans but with relatively early onset. Lesions of interest included the gross enlargement of nerves, particularly those of the brachial plexus, recorded previously for fucosidosis in Springer Spaniels, 5,11 and the many free vacuolated macrophages in the liver with lesser vacuolation of hepatocytes. Perhaps more attention needs to be taken concerning spinal nerves of other LSDs so the specificity, or generality, of this lesion can be established as an aid to provisional diagnosis. The vacuolated macrophages around central veins and in hepatic triads would be expected to actively drain to the hepatic vein traversing to the anterior vena cava and the lymph nodes at the hilum, respectively. 24
The results of this study show the utility of whole-genome sequencing for finding disease-causing mutations, particularly when the pathology is well described and there are a finite number of genes known to be associated with a particular type of disease. Coupled with this mutation’s segregation pattern in tested animals and viewed in relation to the biochemical findings, it confirmed a diagnosis of β-mannosidosis, a disease not previously reported in dogs.
Supplemental Material
Supplemental Material, DS1_VET_10.1177_0300985819839239 - β-Mannosidosis in German Shepherd Dogs
Supplemental Material, DS1_VET_10.1177_0300985819839239 for β-Mannosidosis in German Shepherd Dogs by Robert D. Jolly, Keren E. Dittmer, Dorian J. Garrick, Anastasia Chernyavtseva, Kim M. Hemsley, Barbara King, Michael Fietz, Nicola M. Shackleton, Robert Fairley and Kirsten Wylie in Veterinary Pathology
Supplemental Material
Supplemental Material, DS2_VET_10.1177_0300985819839239 - β-Mannosidosis in German Shepherd Dogs
Supplemental Material, DS2_VET_10.1177_0300985819839239 for β-Mannosidosis in German Shepherd Dogs by Robert D. Jolly, Keren E. Dittmer, Dorian J. Garrick, Anastasia Chernyavtseva, Kim M. Hemsley, Barbara King, Michael Fietz, Nicola M. Shackleton, Robert Fairley and Kirsten Wylie in Veterinary Pathology
Footnotes
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.