
Abstract
Canine cutaneous mast cell tumor (MCT) is the most common canine skin tumor and exhibits variable biologic behavior. Signaling through the KIT receptor tyrosine kinase promotes cellular proliferation and survival and has been shown to play a role in MCT progression. Despite investigations into numerous biomarkers and the proposal of several grading schemas, no single marker or grading system can accurately predict outcome in canine MCT. The first aim of this study was to develop an immunohistochemical assay to measure phosphorylated KIT (pKIT) to investigate its association with 2 commonly used grading systems and other established prognostic markers for canine MCT. Thirty-four archived MCTs were evaluated for expression of pKIT and Ki-67, KIT localization, mitotic count, mutations in exons 8 and 11 in c-kit, and grading by the Patnaik and 2-tier systems. Expression of pKIT was significantly (P < .05) correlated with the 2-tier grading scheme and c-kit mutation. Correlation approached significance (P = .06) with Mitotic Index (MI) and Ki-67. An additional aim was to determine whether pKIT labeling provides a pharmacodynamic marker for predicting response to the receptor tyrosine kinase inhibitor toceranib (TOC). MCTs from 4 of 7 patients demonstrated a partial response to TOC. pKIT expression was assessed by immunohistochemistry in biopsies obtained before and 6 hours after the patients were treated with TOC. Reduced pKIT expression after TOC treatment was demonstrated in 3 of the 4 patients with a partial response compared to 1 of the 3 nonresponders. Collectively, these results demonstrate that immunohistochemical detection of pKIT may be a clinically relevant assay to evaluate the activation status of the major oncogenic pathway in canine MCT.
Mast cell tumors (MCTs) are the most common cutaneous tumors in dogs, accounting for up to 21% of all canine cutaneous tumors, and exhibit extremely variable biologic behavior. 10,12,28 MCT behavior is partially dependent on aberrant signaling of certain proteins, which cannot be detected by routine histopathological evaluation. Activating mutations in the juxtamembrane, kinase, and ligand binding domains of the c-kit proto-oncogene have been associated with the pathogenesis and aggressiveness of canine MCTs, resulting in growth factor–independent constitutive phosphorylation of the KIT receptor tyrosine kinase. 7,11,23,28 Approximately 30% to 50% of histologically intermediate- and high-grade canine MCTs carry a c-kit mutation. 12,28 Our laboratory and others have shown that c-kit mutations, particularly internal tandem duplications in the juxtamembrane domain of exon 11, are associated with an increased incidence of recurrent disease, metastasis, and death. 3,12,20,28 While most gain-of-function mutations of c-kit have been identified in exon 11 of canine MCTs, exons 8 and 9 and, less commonly, exon 17 also acquire activating mutations. 11 Differential labeling patterns of KIT by immunohistochemistry (IHC) in MCTs, associated with mutations in the c-kit proto-oncogene, are indicators of biologic behavior. 10,23 MCTs exhibiting benign biologic behavior tend to demonstrate membrane-associated KIT expression while MCTs associated with aggressive biologic behavior are more likely to demonstrate a redistribution of KIT expression to the cytoplasm. 10 In addition, markers of cellular proliferation such as Ki-67 and AgNOR have been significantly associated with progression of canine MCT. 21,24 These studies demonstrate that markers of proliferation, c-kit mutation status, and KIT protein localization are useful markers of tumor aggressiveness in canine MCT.
Despite this progress in prognostic indicators for canine MCT, additional molecular markers need to be evaluated to further identify the subset of MCTs that are histologically low or intermediate grade but biologically high grade. To date, there are no reports that have examined the relationship between KIT activation status and clinicopathologic features in MCT. The aim of the current study was to retrospectively investigate the relationship between activated KIT expression (phosphorylated KIT [pKIT]) and proliferation indices, KIT localization, c-kit mutation status, and grade in canine MCT by developing an IHC-based assay for pKIT for use in formalin-fixed, paraffin-embedded (FFPE) tumor samples.
In addition to its prognostic impact, the aberrant expression of proteins intimately involved in the tumorigenesis of various neoplasms may also serve as a therapeutic target. As the above studies suggest, KIT plays a significant role in MCT progression and survival. Therefore, small-molecule inhibitors of KIT are an attractive therapeutic strategy for MCTs in dogs. Toceranib (TOC) phosphate (Palladia) is one such inhibitor of KIT that has biological activity against canine MCTs. Indeed, a reduction in pKIT has been demonstrated by Western blot analysis in MCT 8 hours after treatment with TOC compared to pretreatment biopsies. 16 A subsequent clinical trial found that approximately 40% of dogs experienced an objective response to TOC while the remaining 60% did not respond. 13 Therefore, there exists a subpopulation of dogs that will not benefit from TOC treatment. An additional aim of this study was to apply the IHC-based assay for pKIT as a clinically relevant analytical method for predicting response to TOC in canine MCTs to discriminate patients that may respond to TOC from those that are refractory and might benefit from an alternative treatment.
Materials and Methods
Tissue Collection
For the retrospective analysis of KIT activation by pKIT labeling compared to established prognostic parameters for MCT, a total of 34 FFPE primary cutaneous MCTs submitted for MCT profiles were selected from the Colorado State University Investigational Pathology Laboratory archives. For monitoring response or resistance of MCT to TOC, 7 client-owned dogs presenting to the CSU Animal Cancer Center for MCT were enrolled in a clinical trial. Six-millimeter punch biopsies were obtained prior to the first dose (2.75 mg/kg) of TOC (t0) and 6 hours following TOC (t6). The latter falls within the Tmax range for toceranib of 5.3 hours and 9.3 hours. 27 Pryer and coworkers 16 similarly measured levels of pKIT by Western blot 8 hours after administration of toceranib. All dogs received at least 72 hours of pretreatment with prednisone (1 mg/kg every other day [EOD]), diphenhydramine, and omeprazole prior to their biopsy and first toceranib dose. Dog No. 1 received 50 mg/m2 lomustine (CCNU). Tissue was fixed in 10% neutral buffered formalin for less than 48 hours and processed routinely. The 7 dogs described in this study were enrolled in a prospective clinical trial comparing vinblastine and toceranib for the treatment of measurable mast cell disease. This study was performed with approval of Colorado State University’s Institutional Animal Care and Use Committee and with signed informed consent from owners. The 7 dogs enrolled in the prospective study were staged with regional lymph node cytology and abdominal ultrasound (± thoracic radiographs) prior to entry, and those dogs with measurable disease assessable by imaging only underwent reimaging 5 weeks after treatment initiation.
Analysis of Canine MCT Biopsies
Slides were evaluated using light microscopy by 2 board-certified veterinary pathologists (C.H.C.H. and E.J.E.). MCTs from the archived samples were graded by both the Patnaik and 2-tier grading schema. 9,15 The Mitotic Index (MI) was determined on hematoxylin and eosin (HE)–stained slides by counting the number of mitotic figures in 10 consecutive, nonoverlapping 400× fields starting with an area of high mitotic activity and expressed as the number of mitoses per ten 400× fields. 17 Mitotic counts were performed using an Olympus BX41 (Olympus, Waltham, MA, USA) with an eyepiece diaphragm size (field number) of 26.1 mm.
pKIT Antibody Validation
Immunohistochemical detection of activated KIT with a pKIT antibody was validated by Western blot analysis and FFPE HistoGel-embedded cell pellets of c-kit–mutant C2 mastocytoma cells treated with or without TOC. C2 cells were incubated for 6 or 24 hours with increasing concentrations of TOC (0–100 nM). For Western blot analysis, cells were resuspended in lysis buffer containing 1% Triton X-100, 100 nM sodium orthovanadate, 0.2 mM phenylmethylsulfonyl fluoride, 1 M Tris, 1 M NaCl, and 7× protease inhibitor cocktail (Roche Applied Science, Indianapolis, IN), incubated on ice for 15 minutes and centrifuged for 5 minutes. Protein was separated by sodium dodecyl sulfate polyacrylamide gel electrophoresis on a 6% acrylamide gel and transferred onto a polyvinylidene difluoride membrane. Membranes were blocked for 60 minutes at room temperature in 5% bovine serum albumin. Immunolabeling for KIT was performed using a rabbit polyclonal anti-human antibody (Dako, Carpinteria, CA) at 1:1000, and immunolabeling for pKIT was performed using a rabbit polyclonal anti-human antibody targeting the Tyr721 residue (Bioss, Woburn, MA) at 1:1000 for 16 hours at 4°C, followed by incubation with horseradish peroxidase (HRP)–conjugated anti-rabbit antibody at 1:5000 for 30 minutes at room temperature. Immunoreactive bands were detected using enhanced chemiluminescent reagent (Thermo Scientific, Rockford, IL). Quantification of resulting bands was performed using the densitometric analysis function of ImageJ (National Institutes of Health, Bethesda, MD).
For histogel embedding, HistoGel (Thermo Scientific, Pittsburg, PA) was heated in a microwave on low for 5 to 15 seconds. HistoGel was allowed to cool to 50°C before approximately 250 000 cells were resuspended in 40 μL HistoGel. Cell pellets and HistoGel were placed in 10% neutral buffered formalin at 4°C overnight. The HistoGel “plug” containing the resuspended cell pellet was removed and placed inside a tissue cassette with a sponge. HistoGel “plugs” were processed, embedded, and sectioned as standard histology specimens. Immunohistochemical labeling for pKIT was performed as described below.
Immunohistochemistry
Immunohistochemical labeling of FFPE sections was performed by the following procedures. Sections were deparaffinized and rehydrated in descending concentrations of alcohol and water. Heat-induced epitope retrieval with EDTA buffer (pH 9.0) for 30 minutes was followed by endogenous peroxidase blocking with 3% hydrogen peroxide, nonspecific signal blocking with Sniper Blocking Reagent (Biocare, Concord, CA), and incubation with the primary antibody at 4°C overnight. The primary antibodies used for KIT and pKIT were as described above. The primary antibody used for Ki-67 was a rabbit monoclonal (MIB-1 clone; Dako) at a dilution of 1:50. Slides were incubated in a prediluted, HRP-conjugated secondary antibody (Envision; Dako), and immunoreactive complexes were detected using a 3,3′-diaminobenzidine peroxidase substrate detection kit (Vector Laboratories, Burlingame, CA). Purified rabbit immunoglobulin (Dako) was used as a universal negative control (UNC) antibody to ascertain nonspecific labeling. Slides were counterstained with Mayer’s hematoxylin (Sigma Aldrich, St Louis, MO).
Immunohistochemical Evaluation
The growth fraction was determined by counting the number of neoplastic cells immunolabeling for Ki-67 in a total of 500 cells and expressed as the average number of immunopositive cells per 100 cells, evaluated in 5 randomly selected 400× fields, as previously described. 21 This fraction was categorized as “high” or “low” based on a cutoff value of 10.6% immunopositive neoplastic cells, respectively. 21 In the retrospective cases, varying labeling and intensity patterns were defined as 0, no labeling; 1, diffusely cytoplasmic; 2, faint cytoplasmic to perinuclear stippling; and 3, intense cytoplasmic to perinuclear stippling. In addition, pKIT was categorized as “on” (score of 1–3) or “off” (score of 0). For the clinical trial cases, pre- and post-TOC biopsies were evaluated for differences in pKIT labeling in addition to the pattern/intensity as described above. pKIT labeling was evaluated as a percentage of positive tumor cells (the number of positive tumor cells over the total number of tumor cells). Percent reduction was determined using the following calculation: [(post-TOC pKIT score – pre-TOC pKIT score)/pre-TOC pKIT score] × 100. Response Evaluation Criteria In Solid Tumors (RECIST) criteria were used to assess clinical response to TOC therapy. 14 Response criteria were defined as follows: complete response (CR), resolution of all lesions; partial response (PR), greater than or equal to 30% decrease in sum of diameters of all lesions; progressive disease (PD), greater than or equal to 20% increase in sum of diameters of all lesions or appearance of new lesion; and stable disease (SD), less than 30% decrease or less than 20% increase. 14 Similarly, percent tumor reduction was determined using the following calculation: [(best response – baseline)/baseline] × 100. KIT localization and scoring were determined as previously described. 10 Briefly, the KIT- labeling patterns were identified as pattern I, membrane-associated labeling; pattern II, focal to stippled cytoplasmic labeling with decreased or loss of membrane-associated labeling; and pattern III, diffuse cytoplasmic labeling. A percentage of each labeling pattern was determined for each MCT. A final pattern, I, II, or III, was assigned based on the predominant labeling present.
c-kit Mutation Analysis
Mutational analysis for internal tandem duplications in exons 8 and 11 of c-kit was performed using primers designed to amplify the areas of reported mutation (Suppl. Table S1). Together, these 2 primer pairs detect 80% of the activating mutations reported in canine c-kit. 3,5,11 DNA was extracted from tissue within 2 to 3 sections cut from paraffin-embedded blocks (KAPA Express Extract kit, Kapa Biosystems, Wilmington, MA, USA) according to the manufacturer’s instructions. The polymerase chain reaction (PCR) reaction was performed using a kit (Phusion Blood Direct PCR kit; Thermo Fisher, Waltham, MA, USA) in which the Phusion polymerase was designed to be resistant to inhibitors of PCR found in mast cells.
Statistics
Spearman’s rank correlation test was used to determine the correlation of pKIT to ordinal MCT profile variables (Patnaik grading, mitotic count, and KIT localization). The Mann-Whitney test was used to compare pKIT and categorical profile variables (2-tier grading, MI >5/<5, Ki-67, and c-kit mutation status). A P value of <.05 was considered significant.
Results
Grade and Proliferation Indices
By Patnaik grading, 4 of 34 cases were grade 1, 26 of 34 were grade 2, and 4 of 34 were grade 3. By the 2-tier grading scheme, 26 of 34 cases were low while 8 of 34 were high. Mitotic counts ranged from 0 to 25 per ten 400× fields. Using a cutoff value of 10.6 for Ki-67 labeling, 23 of 34 cases had a Ki-67 index of <10.6 while 11 of 34 cases had a Ki-67 index of >10.6. MCTs of the 7 dogs enrolled prospectively were all grade 2.
pKIT Antibody Validation
To determine if the anti-human pKIT antibody could detect canine pKIT, TOC-sensitive exon 11–mutant C2 cells were treated for 24 hours with increasing concentrations of TOC. The anti-pKIT antibody detected a dose-dependent decrease in pKIT relative to KIT and tubulin via Western blot in TOC-treated cells (Figs. 1, 2). Interestingly, total KIT showed a slight downregulation following TOC exposure as well. We postulate this is due to receptor internalization and recycling off of the plasma membrane following inhibition of signaling.
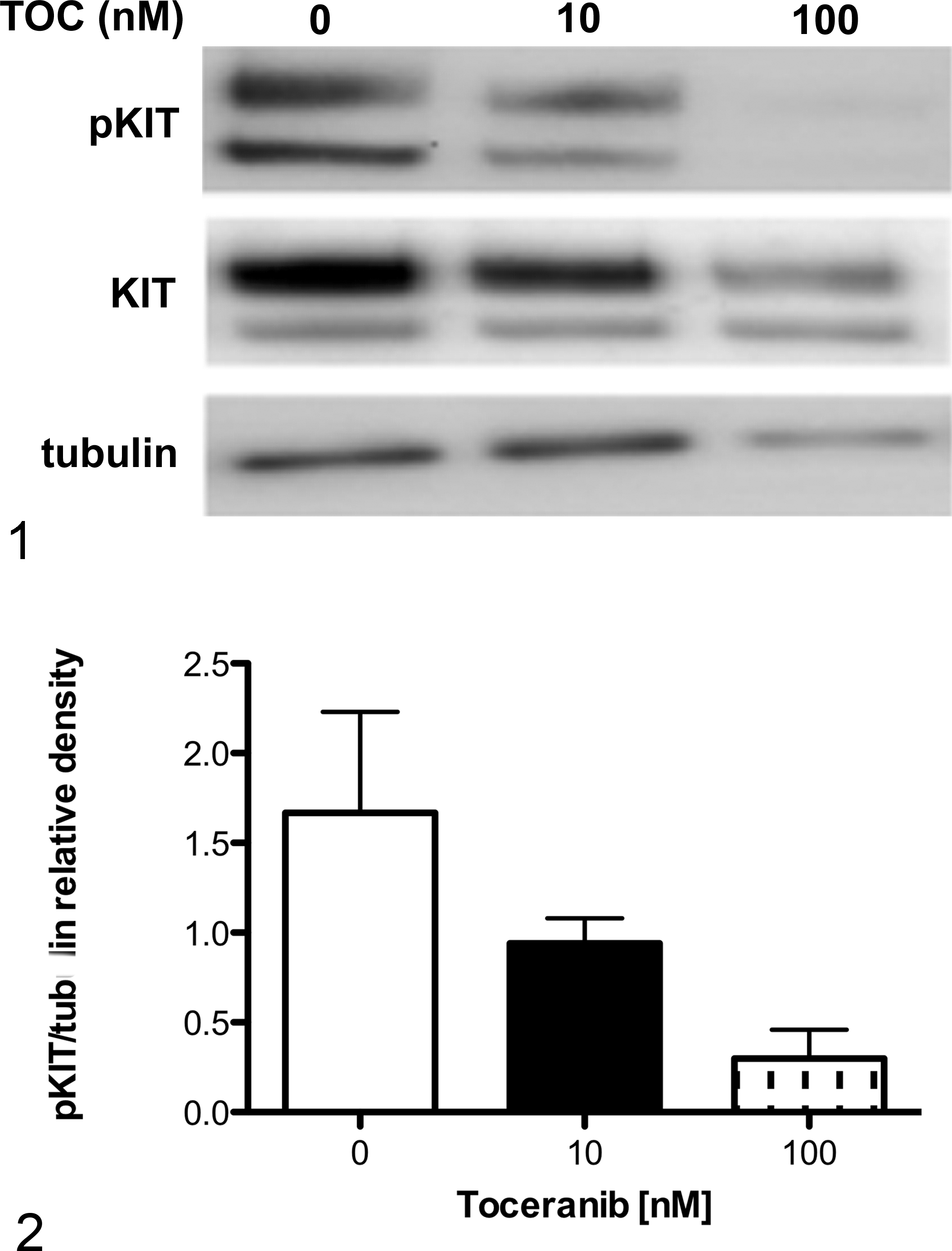
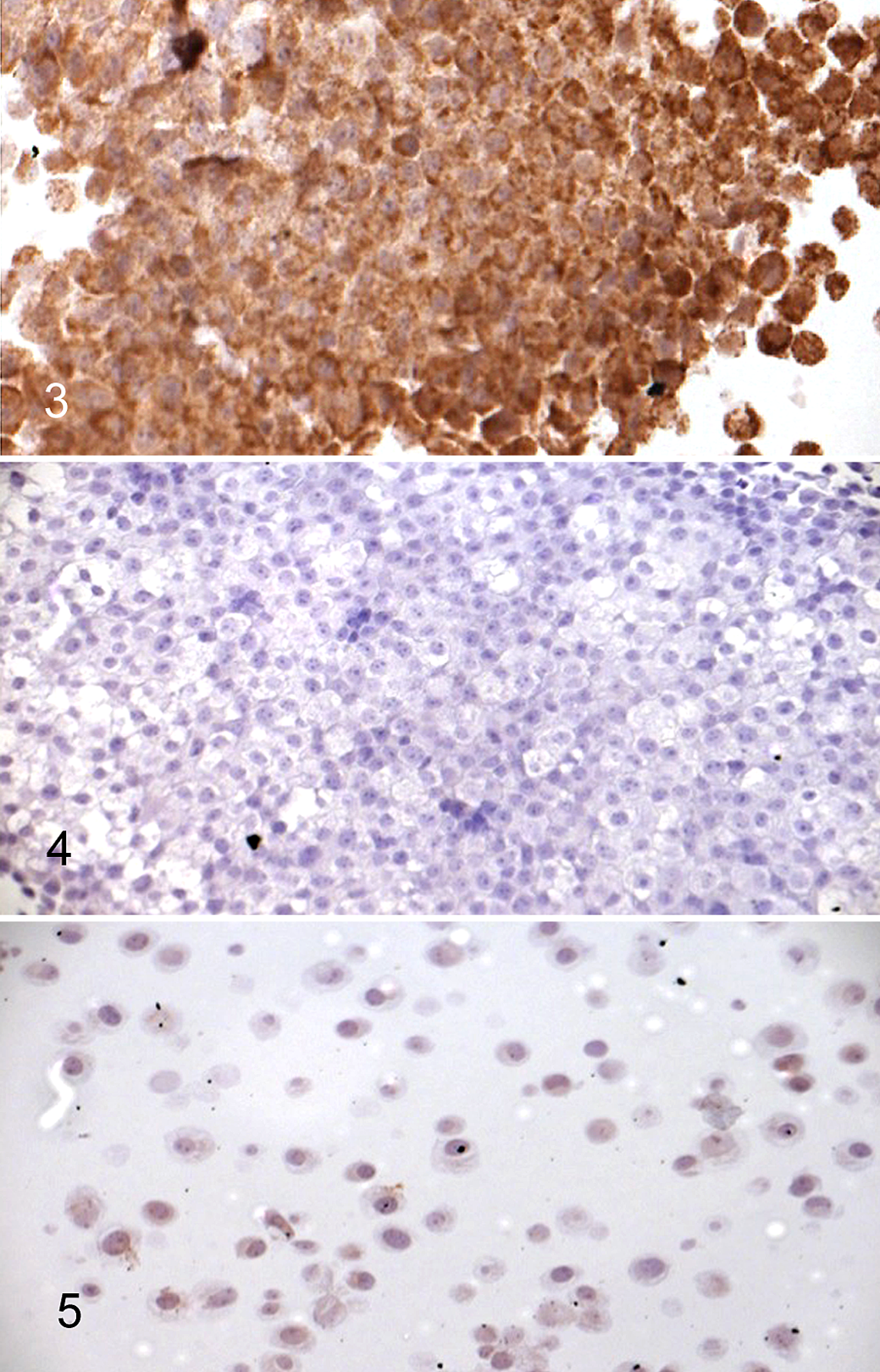
Similarly, to investigate the use of the anti-pKIT antibody in FFPE sections, treated (100 nM TOC) and untreated C2 cells were embedded in HistoGel, processed as standard histologic specimens, and immunostained for pKIT. Untreated cells demonstrated diffuse and intense cytoplasmic immunoreactivity, suggesting widespread activation of the KIT receptor (Fig. 3). This activation, however, was inhibited after treatment of the cells with 100 nM TOC for 6 and 24 hours (Figs. 4, 5), as demonstrated by a decrease in the staining intensity and percentage of cells immunopositive for pKIT. The density of viable cells decreased dramatically after 24 hours of TOC exposure.
pKIT Labeling of Tumor Sections
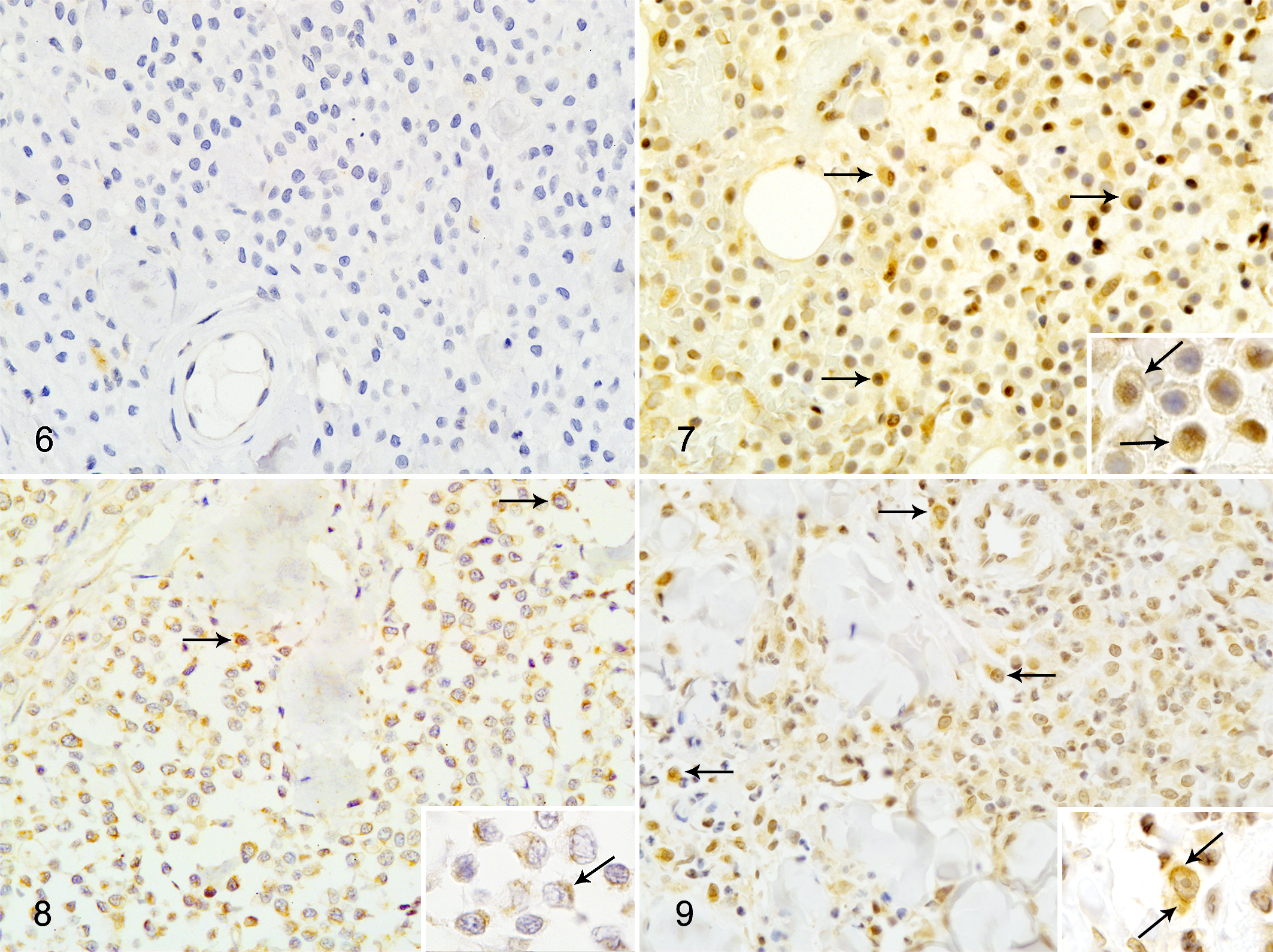
One representative slide from each case (archived and clinical trial cases) was reviewed by 2 board-certified veterinary pathologists (C.H.C.H. and E.J.E.). pKIT labeling was localized to the cytoplasm in 3 patterns (Figs. 6–9): diffuse cytoplasmic labeling (Fig. 7), stippled to globular cytoplasmic labeling of low to moderate intensity (Fig. 8), or high intensity with both cytoplasmic globular and perinuclear labeling (Fig. 9, inset and arrows).
pKIT Expression Correlates With Mitotic Count, Ki-67 Labeling, Mutations in Exon 8 or 11, and 2-Tier Grade
To determine if direct measurement of the activated form of the KIT receptor correlated with histologic grade and other known prognostic factors for MCT, we examined 34 archived canine MCTs for pKIT expression with IHC. Correlation coefficients for the MCT pKIT scores and MCT profile parameters are summarized in Supplemental Table S2. pKIT immunoreactivity correlated significantly with grade as determined using the 2-tier grading system (P = .0157) and c-kit mutation status in exons 8 and 11 (P = .0021). The correlation of pKIT immunoreactivity with MI (P = .065) and Ki-67 growth fraction (P = .065) approached significance.
pKIT as a Marker for Target Modulation
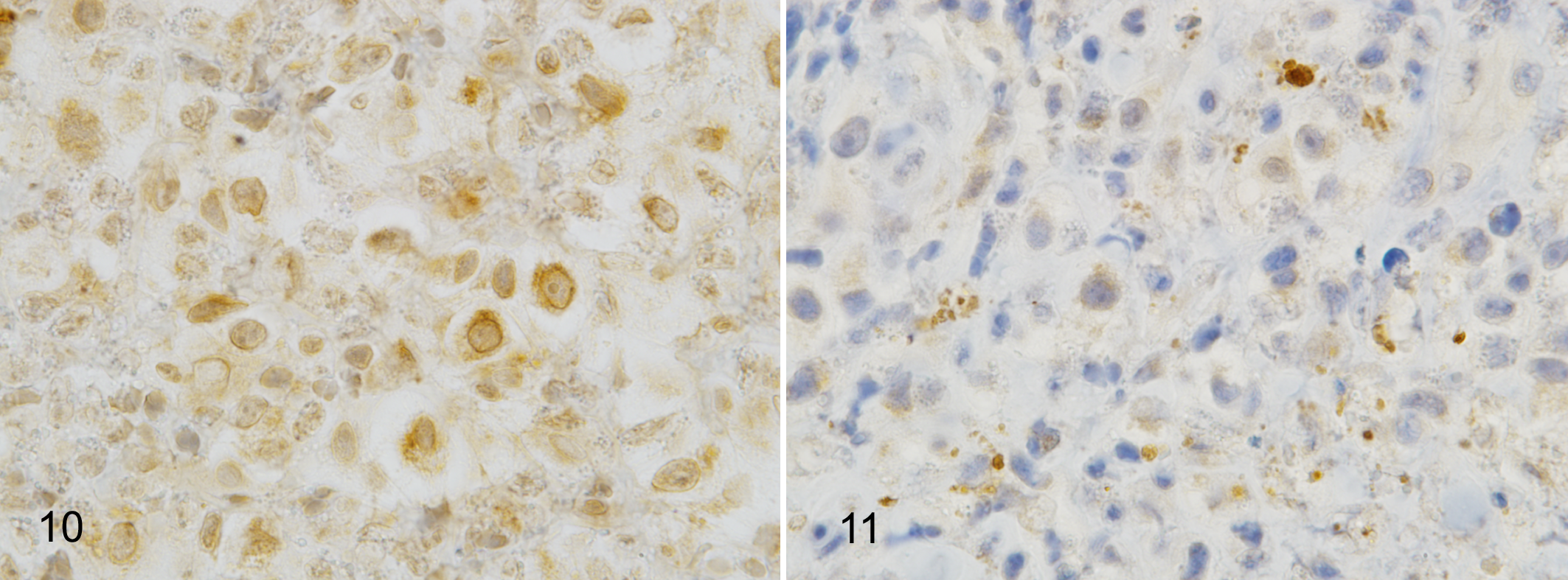
Treatment of dogs with MCT with TOC resulted in a decrease in KIT phosphorylation in the neoplastic cells after 6 hours in 5 of 7 dogs (Table 1; Figs. 10, 11). The remaining 2 of 7 dogs demonstrated either no change (dog No. 3) or a slight increase (dog No. 1) in pre- vs post-TOC pKIT activity by immunohistochemical analysis. MCTs from 4 of 7 patients (dog Nos. 1, 2, 5, and 7) demonstrated a partial response to TOC therapy, 2 of 7 patients (dog Nos. 3 and 6) experienced stable disease, and 1 patient (dog No. 4) experienced progressive disease. Of the 4 patients experiencing partial response, 3 of 4 (75%) demonstrated a reduction in pKIT 6 hours after the first dose of TOC. The fourth patient (dog No. 1) received lomustine 72 hours following TOC initiation; therefore, it is difficult to attribute any reduction of tumor size exclusively to KIT inhibition as lomustine is an effective cytotoxic therapy for MCT. Of the 2 patients that were classified with stable disease, dog No. 3 showed no change in pre- and post-TOC pKIT activity while dog No. 6 demonstrated a 100% reduction in pKIT activity. Finally, 1 patient (dog No. 4) was classified as having progressive disease despite an initial response to therapy as determined by a decrease in pKIT 6 hours post-TOC. However, this patient was removed from the study at the owner’s request after 2 weeks. The patient was diagnosed with multiple cutaneous MCTs. The sum of the diameter of his target lesions was 10.9 cm at day 0, 12.6 cm at 1 week, and 13.2 cm at week 2, a 20% increase compared to baseline. In addition, the patient developed multiple new masses at the 2-week visit. While this patient may have had an initial response to TOC therapy, he later became refractory. Although patient numbers were low, there was no correlation between pre-TOC pKIT score and clinical stage (r 2 = 0.274; P = .278).
Effect of In Vivo Toceranib Treatment (TOC) on Phosphorylated KIT (pKIT) Expression in Canine Mast Cell Tumors.a
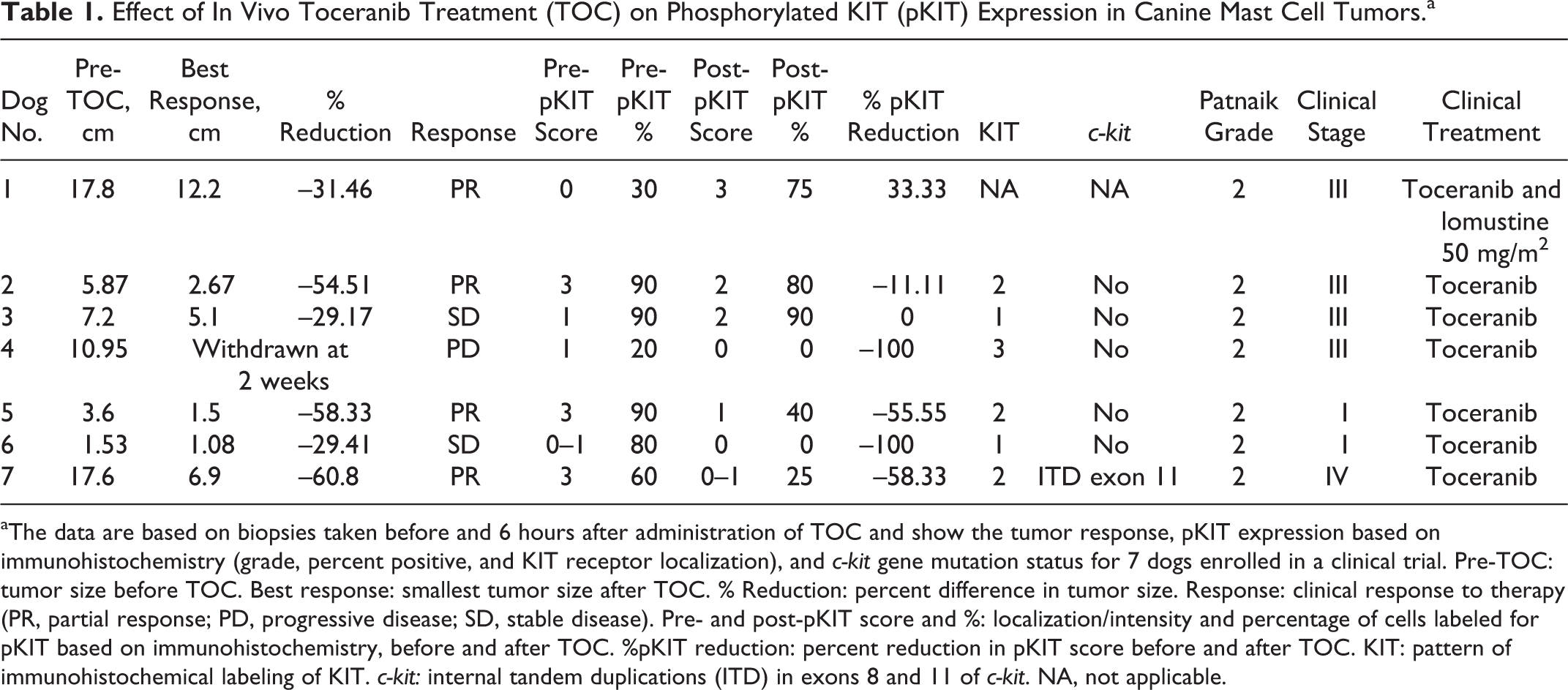
aThe data are based on biopsies taken before and 6 hours after administration of TOC and show the tumor response, pKIT expression based on immunohistochemistry (grade, percent positive, and KIT receptor localization), and c-kit gene mutation status for 7 dogs enrolled in a clinical trial. Pre-TOC: tumor size before TOC. Best response: smallest tumor size after TOC. % Reduction: percent difference in tumor size. Response: clinical response to therapy (PR, partial response; PD, progressive disease; SD, stable disease). Pre- and post-pKIT score and %: localization/intensity and percentage of cells labeled for pKIT based on immunohistochemistry, before and after TOC. %pKIT reduction: percent reduction in pKIT score before and after TOC. KIT: pattern of immunohistochemical labeling of KIT. c-kit: internal tandem duplications (ITD) in exons 8 and 11 of c-kit. NA, not applicable.
Discussion
The aims of this study were to investigate the relationship between activated KIT expression (pKIT) and established prognostic variables for canine MCT by establishing a reliable and clinically applicable immunohistochemical assay. In addition, immunohistochemical labeling of MCT for pKIT might offer an effective method to monitor response to the KIT inhibitor, toceranib.
To the authors’ knowledge, this is the first report of the IHC detection of pKIT using a phosphospecific antibody in FFPE canine MCTs. We have applied this IHC-based assay of KIT activation to explore its correlation to other established prognostic parameters in canine MCT as well as monitoring tumor response to inhibitors of KIT.
To validate the specificity of the selected pKIT antibody, we used the previously established C2 mastocytoma cell line. This cell line was established from a spontaneously occurring canine MCT and harbors the commonly reported internal tandem duplication in exon 11 of the c-kit gene. 6 As a result of this mutation, the KIT receptor in C2 cells is constitutively phosphorylated. The anti-pKIT antibody recognized canine pKIT by both Western analysis of C2 cell lysates and cytoplasmic IHC labeling of C2 cells embedded in HistoGel. Furthermore, upon treatment of C2 cells with TOC, pKIT immunoreactivity was decreased by both Western analysis and IHC. This demonstrated the specificity and potential applicability of this antibody to investigate KIT activation in canine MCTs. We subsequently used this pKIT antibody for 2 applications: (1) to retrospectively investigate the correlation of activated KIT in MCT to other established prognostic parameters that comprise the MCT profile offered through the Investigational Pathology Lab at CSU and (2) to monitor the pharmacodynamic response to TOC in client-owned animals presenting to CSU for the treatment of MCT.
Current prognostic parameters for canine MCT include grade, 9,15 mitotic count, 17 KIT localization, 10,23 Ki-67, 21 c-kit mutation status, 3,7,12 and clinical staging. 19,22 A review of these parameters is beyond the scope of this study; however, while they can be used to interpret the activation status of KIT, they do not provide a direct measurement of the activated receptor. In the current study, there was a significant or near-significant correlation between pKIT labeling and MI, Ki-67, c-kit mutation status, and grade by the 2-tier scheme. Inclusion of additional cases would provide increased statistical power to these analyses. Upon activation, receptor tyrosine kinases are rapidly internalized and recycled off the plasma membrane. 25 Constitutively active KIT due to mutations in c-kit, as well as subsequent rapid receptor internalization, is likely the reason for the loss of membranous labeling and increased cytoplasmic labeling in biologically aggressive MCTs. Intracellular, not membranous, localization of mutant KIT is responsible for downstream oncogenic signaling. 26 For further scrutiny of these findings, KIT labeling was redefined as either membranous or cytoplasmic since less aggressive MCT cells demonstrate KIT expression limited to the membrane and more aggressive MCT cells display a redistribution of KIT to the cytoplasm with loss of membranous labeling. 10 Following this redefinition, correlation between pKIT and KIT redistribution (membranous or cytoplasmic) again neared significance (P = .105).
Neither c-kit mutations nor a redistribution of KIT were identified in a small subset of tumors in this study. Internal tandem duplication mutations in exons 8 and 11 were examined in this study. In addition, other mutations in c-kit, such as activating mutations in exons 9 and 17, could contribute to the positive labeling for pKIT. Furthermore, alternative mechanisms of KIT activation other than direct mutation of the KIT receptor have been reported. The KIT receptor ligand, stem cell factor (SCF), has recently been shown to be overexpressed in canine MCT independent of activating mutations in KIT. Production of SCF was demonstrated in Ki-67–positive MCT by immunohistochemistry. This suggests that autocrine/paracrine production of SCF also contributes to the growth and survival of canine MCT. 1,2
In addition to biomarkers, histologic grade has been widely and more traditionally used as an indicator of biologic behavior in MCT. Histologic grading by the Patnaik system has been the gold standard and has provided a strong foundation in the grading of canine cutaneous MCTs, but certain criteria within the grading system require subjective interpretation, and significant interpathologist variability exists. 15 An additional limitation of the Patnaik grading system has been the frequency at which grade 2 MCTs are diagnosed and further complicated by the observation that some grade 2 MCTs are fairly benign while others are biologically aggressive. The 2-tier system attempts to address the predominance of Patnaik grade 2 MCTs and the ambiguity and biologic variability within this group. 9 Since its introduction, grading by the 2-tier system has been independently validated by several groups. 18,20,21 Results of these studies highlight the significantly higher intraobserver concordance and prognostication of the 2-tier system compared to the Patnaik system. Interestingly, in the current study, expression of pKIT was significantly correlated to grade by the 2-tier system while no significant correlation was shown between pKIT and Patnaik grade. However, when Patnaik grades 1 and 2 were grouped together, there was a highly significant correlation (r = 0.41; P = .007) between pKIT and grades 1/2 and grade 3. Recently, 2 independent studies comparing the utility of Patnaik and 2-tier grading schemes concluded that there were no significant prognostic differences between Patnaik grades 1 and 2. 18,20 Overall, these results suggest that expression of pKIT correlates significantly with other commonly used indicators of aggressiveness, such as grade, in canine MCT.
The activated KIT receptor represents a viable therapeutic target for the treatment of canine MCT. As such, targeted inhibitors of KIT have been developed for the treatment of recurrent, nonresectable MCTs and have shown clinical efficacy, particularly in tumors demonstrating mutations in the KIT receptor. 13,16 However, de novo and acquired resistance to targeted therapy remains a significant clinical challenge. 4,8 Reproducible and clinically relevant assays are needed to identify patients most likely to respond to targeted therapy as well as those that are refractory to such treatment. We have developed an IHC-based assay to measure changes in activated KIT as an indication of target modulation. Others similarly measured response to TOC in dogs with MCT by assessing pre- and posttreatment pKIT levels via Western analysis. While this is a reasonable approach, Western analysis is time-consuming and requires adequate tumor tissue sample size collected under specific conditions. In contrast, tissue for IHC evaluation is collected by routine, clinically relevant procedures and requires less tissue. In addition, any potential contribution of signal from the tumor stroma can be visually excluded by immunohistochemical evaluation. This is in contrast to Western analysis, in which whole tissue lysates are evaluated. Regardless of the method used, valid biomarkers are needed to effectively monitor response to treatment and, more important, identify patients unlikely to respond. Early identification of treatment failure is critical to adjusting the treatment plan so that these patients may benefit from second-generation tyrosine kinase inhibitors or alternative therapies. We have shown that immunohistochemical detection of pKIT prior to and 6 hours after TOC is a way by which response to TOC in canine MCT could be monitored, to detect reduced expression of pKIT in response to TOC therapy. Although there are practical limitations to re-biopsying a patient, serial fine-needle aspirates could be used to similarly monitor KIT activation. Correlation of tumor response and reduction of pKIT were not statistically significant, most likely due to the limited number of patients enrolled. However, the trend suggests that in patients demonstrating a partial response to TOC, there is reduction of KIT activation by immunohistochemical analysis of pKIT. These preliminary data justify further investigation of pKIT modulation as a predictive factor in TOC-treated canine MCT patients.
Collectively, these results demonstrate that IHC detection of pKIT may be a clinically relevant assay to evaluate the activation status of the major oncogenic pathway in canine MCT. As such, it may serve as an indication of the aggressiveness of the tumor as well as a rapid pharmacodynamic biomarker that demonstrates successful or unsuccessful target modulation. Future studies should be performed to assess the prognostic significance of pKIT expression in a cohort of uniformly treated and systematically evaluated canine patients with MCT.
Footnotes
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by Colorado State University College Research Council Grant 1682002 and American Kennel Club Canine Health Foundation Grant 01426. Toceranib active pharmaceutical ingredient and Palladia capsules were provided by Zoetis, Inc.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.