
Abstract
Domoic acid (DA) is a neurotoxin reported to produce damage to the hippocampus, which plays an important role in memory. The authors inoculated rats intraperitoneally with an effective toxic dose of DA to study the distribution of the toxin in major internal organs by using immunohistochemistry, as well as to evaluate the induced pathology by means of histopathologic and immunohistochemical methods at different time points after toxin administration (6, 10, and 24 hours; 5 and 54 days). DA was detected by immunohistochemistry exclusively in pyramidal neurons of the hippocampus at 6 and 10 hours after dosing. Lesions induced by DA were prominent at 5 days following treatment in selected regions of the brain: hippocampus, amygdala, piriform and perirhinal cortices, olfactory tubercle, septal nuclei, and thalamus. The authors found 2 types of lesions: delayed death of selective neurons and large areas of necrosis, both accompanied by astrocytosis and microgliosis. At 54 days after DA exposure, the pathology was characterized by still-distinguishable dying neurons, calcified lesions in the thalamus, persistent astrocytosis, and pronounced microgliosis. The expression of nitric oxide synthases suggests a role for nitric oxide in the pathogenesis of neuronal degeneration and chronic inflammation induced by DA in the brain.
Domoic acid (DA) is a naturally occurring excitotoxin produced by the marine red alga Chondria armata and by various subspecies of marine diatoms of the genera Nitzschia and Pseudonitzschia. 8,28 Under certain environmental conditions, DA-producing species grow in an exponential fashion, and the toxin accumulates in filter-feeding species and fish, such as crabs, razor clams, scallops, mussels, squids, anchovies, and sardines. 7,11,31 Once transferred to the food chain, DA can be bioconcentrated and may cause poisoning of marine mammals, seabirds, and humans. 30,35,41
DA poisoning was first reported in Canada in 1987. More than 143 people became ill and 4 elderly people died after consuming cultivated blue mussels (Mytilus edulis) harvested in the Cardigan Bay of Prince Edward Island. 8,36,55 The affected people presented gastrointestinal signs (nausea, vomiting, abdominal cramps, diarrhea), cardiovascular signs (unstable blood pressure, cardiac arrhythmias), and neurologic signs (disorientation, confusion, coma, seizures, memory loss). As many of the affected survivors showed permanent short-term memory loss, the intoxication was termed amnesic shellfish poisoning. 36 After this incident, programs to monitor DA presence in water and concentration in shellfish were implemented in many countries. 34
Histologic examination of deceased people showed brain damage characterized by neuronal loss and astrocytosis, predominantly involving hippocampus and amygdala, but lesions were also found in other brain structures, such as thalamus and prefrontal cortex. 48 Lesions induced by DA in the brain of animal models have a similar pattern in terms of location and nature, most notably involving the hippocampus and amygdala, but histologic changes can also be found in secondary olfactory areas, lateral septum, thalamus, or area postrema, among other nervous structures. 36 The neuropathology is characterized in humans and animals by neuronal loss and reactive gliosis. Importantly, neuronal degeneration can be detected only from 24 hours onward, being most severe at 5 to 14 days following DA exposure. 2 But there are important differences in species susceptibility, as humans appear to be much more sensitive to the acute oral toxicity of DA than nonhuman primates, whereas rodents are less sensitive, with rats being more resistant to poisoning than mice after oral administration. 30 Irrespective of the route of administration, DA intoxication is evidenced by behavioral indicators such as sluggishness, stereotypic scratching, profuse salivation, tremor, seizure, and death. 5
DA is a water-soluble tricarboxylic acid, with a secondary amino group, and structurally very similar to another known neurotoxin, kainic acid (KA). 15 DA and KA are analogues of the excitatory neurotransmitter glutamate, and current evidence supports the view that DA and KA interact and activate glutamate receptors (GluRs) to elicit a potent excitotoxic response that leads to neuronal cell death. More precisely, DA neurotoxicity is mediated by its interaction with alpha-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) and KA ionotropic GluRs. Activation of AMPA/KA receptors induces a massive influx of calcium ions (Ca2+) into neurons, resulting in the release of glutamate, which in turn activates N-methyl-D-aspartate receptors that promote further glutamate release and excitotoxicity. 24 The elevated levels of intracellular Ca2+ also activate Ca2+-dependent enzyme systems and signaling cascades resulting in exaggerated free radical generation and oxidative and nitrosative stress, as well as activation of phospholipases, protein kinase C, proteases, protein phosphatases, nitric oxide synthases (NOSs), and caspases. 23,29,36 DA might also activate GluRs outside the central nervous system—that is, in peripheral organs expressing GluRs, such as kidneys, liver, and lungs. 22
In this study, rats were inoculated with various doses of DA and then sacrificed at different time intervals after the exposure with the following objectives: (1) to investigate the presence of the toxin in the main internal organs and (2) to reevaluate the pathology induced by DA in the entire brain and in other internal organs.
Material and Methods
Chemicals
DA (>97%), tomato (Lycopersicon esculentum) lectin, triton X-100, and an anti-MAP-2 antibody were purchased from Sigma-Aldrich (Stainheim, Germany). Tween 20 was provided by Panreac Química SAU (Barcelona, Spain). Dako Real Peroxidase-Blocking Solution, the secondary antibody solution (Dako REALTM EnVision Detection System), diaminobenzidine (DAB+ Chromogen), and the anti–glial fibrillary acidic protein (anti-GFAP) antibody were purchased from Dako (Barcelona, Spain).
DPX was provided by BDH Laboratory Supplies (Poole, United Kingdom). Vectastain ABC reagent was purchased from Vector Laboratories (Peterborough, United Kingdom) and the polyclonal pan-antibody anti–universal NOS by Thermo Scientific (Madrid, Spain).
The monoclonal primary antibody anti-DA was obtained courtesy of Dr Christopher T. Elliott, Institute of Agri-Food and Land Use, School of Biological Sciences, Queen’s University (Belfast, Northern Ireland). This antibody was prepared using a DA-bovine thyroglobulin conjugate in the same manner as that described by Traynor et al. 33,49 With this immunogen, monoclonal antibodies were produced as reported in the fusion procedure by Galfre and Milstein. 20 We have successfully employed the DA monoclonal antibody in our laboratory, showing that it possesses a high affinity for its target. 19
Animals
Thirty-two adult female Sprague-Dawley rats (weighing 180–215 g; mean weight, 200 g) were used. Animals were individually housed, with a rodent diet and water provided ad libitum, and maintained at a controlled temperature (23 ± 2°C) and humidity (60%–70%), under a 12-hour light:dark cycle for 1 week before the experiments. Animals were fed also ad libitum throughout the experimental period.
All the procedures using animals were approved by the Institutional Animal Care Committee of the Universidad de Santiago de Compostela. All possible efforts were made to reduce animal suffering and minimize the number of animals used.
Determination of an Effective Toxic Dose of DA
An experiment was designed to select a dose of DA that, while inducing major behavioral alterations, did not cause the death of animals. Four doses of DA were tested: 1.5, 2, 2.5, and 3.5 mg/kg. Fifteen rats were randomly divided into 5 groups, each of 3 animals. Four groups were inoculated intraperitoneally (ip) with the corresponding dose of DA diluted in physiologic saline solution. The exact dose was calculated per the weight of each animal, and the toxin (from a stock solution of DA at 10 mg/ml in purified water) was diluted to provide the required dose in 0.5 ml. The remaining group was inoculated ip with physiologic saline solution and used as control.
The animals were habituated to an acrylic glass observation chamber for 30 minutes before DA or saline administration. Immediately after inoculation, the animals were returned to the observation chamber, and their behaviors were observed for 6 hours in a cycle of 5 minutes on and 10 minutes off. Control and experimental animals were sacrificed with an ip overdose of sodium pentobarbital. Then they were perfused intracardially with physiologic saline (37°C) containing heparin (0.1% v/v) and sodium nitrate (0.025% v/v), followed by 10% neutral buffered formalin. The encephalon, liver, kidneys, lungs, and heart were removed, immersed in neutral buffered formalin, and embedded in paraffin. Sections obtained from these samples were stained with hematoxylin and eosin (HE) and cresyl fast violet for Nissl substance. 6
The dose of 2.5 mg/kg was selected to study short- and long-term pathologic effects of DA. Rats (n = 17) were injected with 2.5 mg/kg of DA and sacrificed at 6 hours (n = 3), 10 hours (n = 3) and 24 hours (n = 3; short-term effects), as well as at 5 days (n = 3) and 54 days (n = 3; long-term effects). Two animals died 5 to 10 minutes after DA administration and were replaced by 2 new animals. Animals were sacrificed with an ip overdose of sodium pentobarbital and then were perfused intracardially with physiologic saline, followed by Bouin solution. The brain and other internal organs (liver, kidneys, lungs, and heart) were removed and immersed in Bouin solution.
The brain was divided into 5 coronal blocks: (1) lateral olfactory tract, (2) amygdala and hypothalamus, (3) thalamus and head of the hippocampus (4) piriform lobe and tail of the hippocampus, (5) cerebellum and brainstem at the level of the pyramidal tracts. The brain blocks and tissue samples randomly taken from the other internal organs were embedded in paraffin according to standard laboratory procedures. Paraffin-embedded sections were cut 3 μm thick, mounted on silanized slides, and dried overnight at 37°C. Paraffin sections were stained with HE and examined under the light microscope for morphologic analyses. Adjacent sections of the brain were stained with cresyl fast violet for Nissl substance 6 and alizarin red S and von Kossa for histochemical detection of calcium salt deposits.
Immunohistochemical (IHC) assays were done for DA detection in sections from all organs using a monoclonal primary antibody anti-DA. Other antibodies used in brain sections were as follows: a monoclonal anti-microtubule-associated protein 2 (anti-MAP-2) antibody to assess neuronal integrity, a polyclonal anti-GFAP antibody for astrocyte labeling, and, only on hippocampal sections, a polyclonal anti–universal NOS that detects the 3 isoforms of NOS (neuronal, nNOS; endothelial, eNOS; and inducible, iNOS). For all IHC methods, sections were deparaffinized with xylene and rehydrated through a graded alcohol series. To block peroxidase activity and prevent nonspecific staining, 1 hour of pretreatment with Dako Real Peroxidase-Blocking Solution (Dako) was performed. After removal of blocking reagents, sections were rinsed 3 times in phosphate buffered saline (PBS) with 0.005% Tween 20 and then incubated with the primary antibody. Thereafter, sections were rinsed 3 times in PBS-Tween 20 and incubated with the secondary antibody solution during 30 minutes. Samples were washed 3 times with PBS-Tween 20 and revealed with diaminobenzidine. Finally, the slides were counterstained with hematoxylin, dehydrated, and permanently mounted in DPX.
Tomato (L. esculentum) lectin was used for the staining of microglia, as follows. Endogenous peroxidase activity was blocked by incubation with 0.3% hydrogen peroxide in methanol for 15 minutes. After 2 rinses in Tris-buffered saline (TBS) and a 15-minutes pretreatment with 0.3% Triton X-100 in TBS, the sections were incubated in biotin-labeled tomato lectin (10 μg/ml) in TBS with 0.3% of Triton X-100 for 2 hours at room temperature, rinsed 3 times with TBS, and then labeled with Vectastain ABC reagent (1:250 in PBS) for 90 minutes at room temperature. Visualization of peroxidase activity in microglial cells was done by incubation in diaminobenzidine.
To test the specificity of the assays, positive and negative control slides were included in each series of stained sections. For positive controls, we used the following: for DA staining, thigh sections of a rat from a previous experiment that was inoculated intramuscularly with DA in the quadriceps muscle and sacrificed 5 minutes after treatment; for staining of MAP-2, GFAP, and tomato lectin, brain sections from noninoculated control rats; and for the uNOS antibody, sections of human tonsil and sections of lymphoid and nervous tissues from noninoculated control rats. For negative controls, we used slides incubated with buffer instead of antibody or lectin dilutions and, for the DA antibody, sections from nontreated control rats. Antigen retrieval methods, as well as dilution and incubation times of the different primary antibodies and lectin employed in our study, are shown in Table 1.
Concentrations, Incubation Times, and Pretreatment of the Primary Antibodies and Tomato Lectin.

Abbreviations: DA, domoic acid; GFAP, glial fibrillary acidic protein; MAP-2, microtubule-associated protein 2; RT, room temperature; uNOS, universal nitric oxide synthase.
Results
Animals inoculated ip with the lower doses of DA (1.5 and 2 mg/kg) showed reduced activity, minor scratching, and bristly hair coat for 5 to 10 minutes and then resumed normal activity. Animals treated with the highest dose (3.5 mg/kg) became moribund and died 5 to 10 minutes following the injection after showing reduced activity.
Microscopic examination of brain, heart, lungs, liver, and kidneys from animals treated with the lower doses, animals dying immediately after DA administration, and control animals revealed absence of lesions or immunostaining for the toxin.
Animals receiving 2.5 mg/kg of DA displayed clinical signs typical of DA intoxication, such as generalized hyperactivity, head scratching producing eyelid and periocular area abrasions, rigidity, loss of postural control, tremors, and head jerks. These animals that survived DA treatment and displayed neurologic signs were sacrificed at different time points after inoculation (6, 10, and 24 hours; 5 and 54 days).
The brain of rats inoculated with 2.5 mg/kg of DA and sacrificed at 6, 10, and 24 hours after toxin administration showed no histologic changes. However, DA presence was readily detected by immunohistochemistry in the brain at 6 hours after ip inoculation of the toxin. Immunoreactivity was observed exclusively in the perikaryon of pyramidal neurons of CA1, CA2, and CA3 subfields of the hippocampus, with the immunostaining being more intense in neurons of the CA3 subfield (Figs. 1–3, Supplemental Fig. 1). Although reduced in number, at 10 hours, DA-positive neurons were still present in the same areas of the hippocampus but with no differences in intensity of immunostaining among subfields as was apparent at 6 hours. In these animals sacrificed at 6 and 10 hours after toxin administration, immunoreactivity against DA was not observed in other brain areas; at later postinoculation times, neither region of the brain contained DA-positive cells. Positive immunostaining was not found in other internal organs at any of the time points investigated.
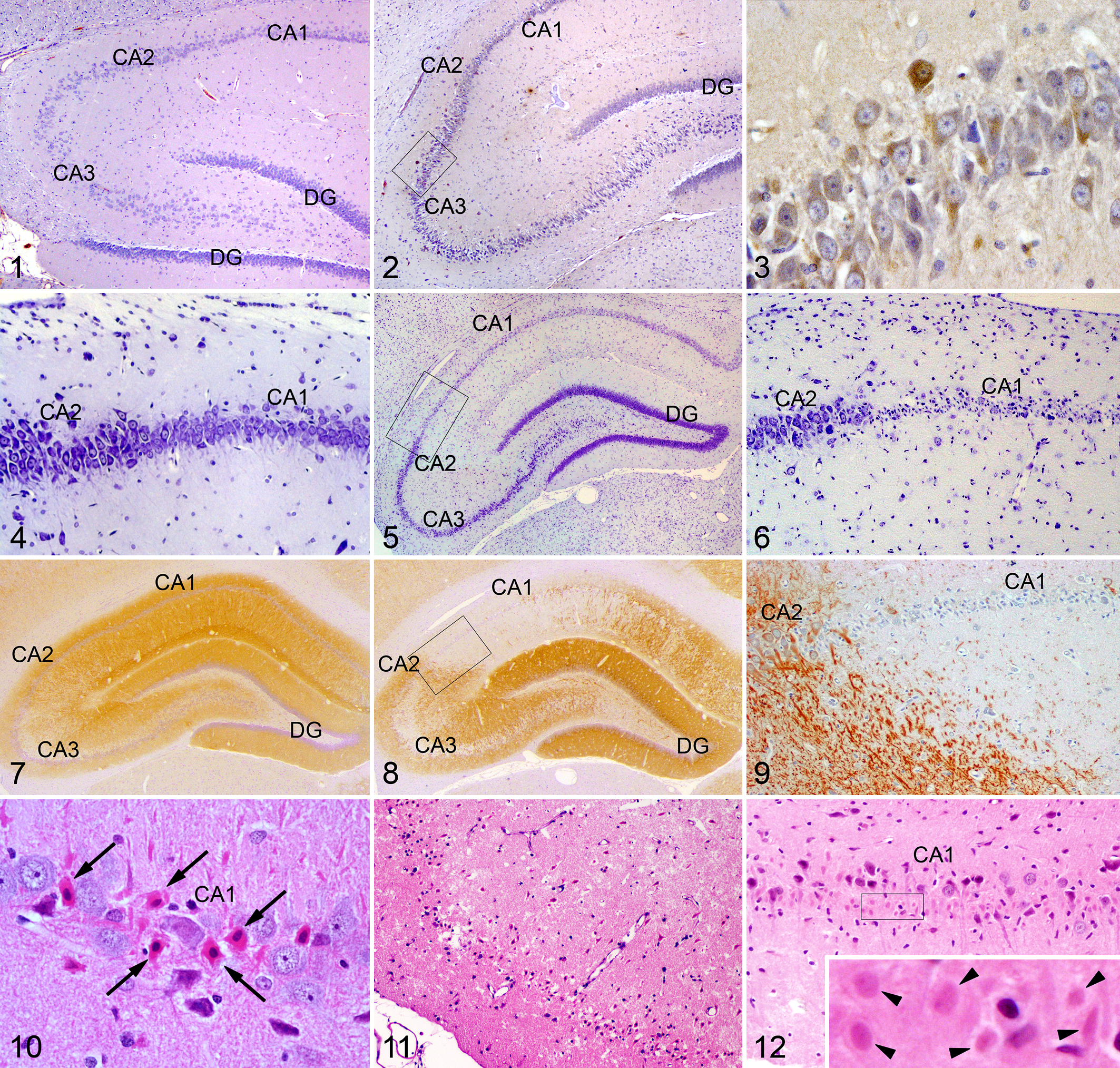
Hippocampus; rat. CA: cornu ammonis; DG: dentate gyrus. Immunohistochemistry for domoic acid (DA).
At 5 days following treatment with 2.5 mg/kg of DA, 2 distinct types of lesions were observed in the brain: selective neuronal degeneration and large areas of tissue damage. The first was characterized by death of neurons—particularly in hippocampus, amygdaloid complex, and piriform and perirhinal cortices but also in olfactory tubercle, lateral septum, and several thalamic nuclei. Neuronal loss was especially remarkable in the hippocampal CA1 and CA3 subfields and hilus of the dentate gyrus, while the CA2 subfield seemed spared (Figs. 4–6).
IHC staining with the antibody anti-MAP-2, a marker of neuronal integrity, identified the extensive loss of neurons in the hippocampus (Figs. 7–9). In sections stained by HE, dying neurons in all damaged areas exhibited ischemic morphology—that is, acute shrinkage with angular profiles, eosinophilia of the cytoplasm, and darkly stained and homogeneous nucleus (Fig. 10).
The second type of lesion found at 5 days after dosing was the occurrence of large, well-defined areas of necrosis in the limbic system, especially in the piriform cortex (Fig. 11) and amygdaloid complex, but small zones were also observed in dorsomedial and dorsolateral regions of the thalamus and in CA1 and CA3 subfields of the hippocampus. Necrotic areas were distinguished from normal tissue as they appeared paler and moth-eaten, due to the extensive vacuolization of the neuropil, and contained shrunken and eosinophilic neurons and other cells with pyknotic nuclei. MAP-2 staining (data not shown) was absent in necrotic areas.
At 54 days after DA treatment at a dose of 2.5 mg/kg, dying neurons were still distinguishable in damaged areas but with a different morphology compared with that observed at 5 days: degenerating neurons were difficult to distinguish from surrounding structures, since they showed undefined outlines and faintly stained and shrunken perikarya and nuclei (ghost neurons; Fig. 12, Supplemental Fig. 3). At 54 days, extensive areas of necrosis were detected in the same structures as at 5 days but showing a more advanced stage of disintegration of normal tissue architecture (Supplemental Figs. 2, 3). In addition, calcification of injured thalamic nuclei was observed in HE-stained sections and confirmed by alizarin red S and von Kossa staining methods (Supplemental Figs. 4–7) in all animals sacrificed at 54 days after DA dosing.
GFAP immunostaining showed an increase in the number and size of astrocytes in hippocampus, piriform and perirhinal cortices, amygdala, and thalamus at 5 days following administration of 2.5 mg/kg of DA, compared with controls (Supplemental Figs. 8, 9, 11, 12). Astrocytosis was still apparent in DA-inoculated animals in the same brain structures at 54 days after toxin treatment (Supplemental Figs. 10, 13). GFAP staining was absent from areas with loss of structure (Figs. 13–15), coincident with the large areas of necrosis in HE-stained slides at 5 and 54 days after dosing.
Tomato lectin histochemistry showed an increase in the number of labeled microglial cells in all affected regions after 5 days and especially 54 days after treatment with 2.5 mg/kg of DA (Figs. 16–18). Microglial cells were especially prominent within and around necrotic areas and in calcified thalamic lesions (Figs. 19–21).
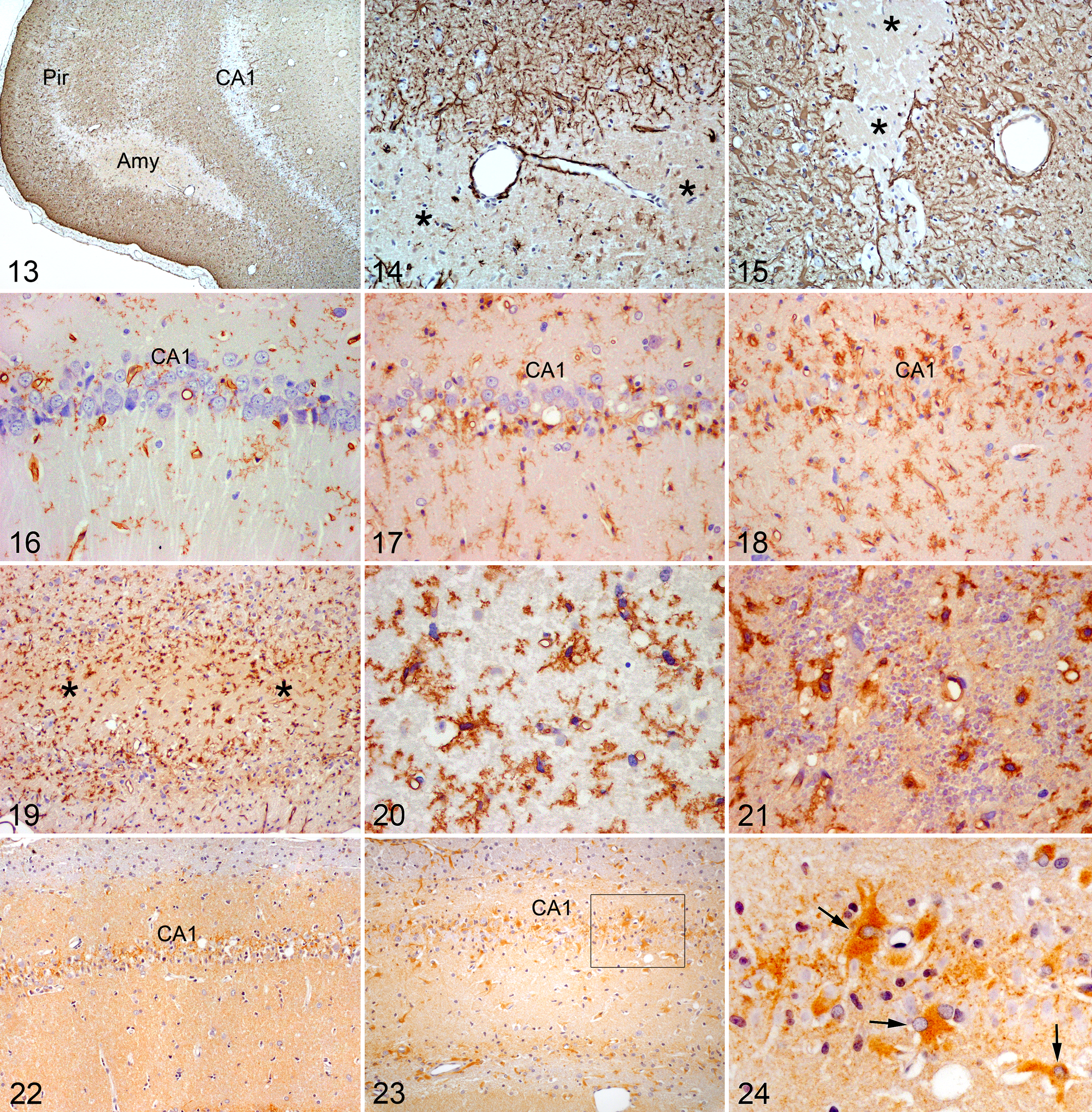
Brain; rat. Immunohistochemistry for glial fibrillary acidic protein (GFAP).
The basal expression of NOS in the hippocampus of the rats used in this study, as exposed by immunohistochemistry via a uNOS antibody, was very low in control animals. At 5 days after DA administration at a dose of 2.5 mg/kg, a slight increase in NOS expression was observed in the pyramidal cell layer and localized mainly in the perikaryon of neurons and probably also in surrounding astrocytes (Fig. 22). At 54 days, the pattern of NOS expression was drastically different, with positive cells showing an intense immunostaining and displaying morphologic and distribution characteristics coincident with those presented by astrocytes in sections processed for GFAP immunohistochemistry (Figs. 23, 24).
Table 2 summarizes the pathologic and IHC findings of the study.
Immunohistochemical and Histopathologic Findings in Rats Inoculated With 2.5 mg/kg of Domoic Acid.a

Abbreviations: DA, domoic acid; uNOS, universal nitric oxide synthase. –, no staining or damage; +/–, basal or normal expression of uNOS; +, mild staining or damage; ++, moderate staining or damage; +++, intense staining or severe damage.
aThere were no significant differences among animals of the same group.
bThe criteria applies to hippocampus.
cThe criteria applies to limbic system.
dThe criteria applies to necrotic thalamic nuclei.
Discussion
In this work, we treated rats systemically with the natural excitotoxin DA to investigate the distribution of the toxin by using immunohistochemistry and the induced pathology at various doses and postinoculation time intervals.
Lower doses of DA (1.5 or 2.0 mg/kg of DA) produced only minor signs in treated animals; alternatively, the highest dose (3.5 mg/kg of DA), which caused rapid death, did not induce histologic changes, and the toxin was not detected by immunohistochemistry in any organ at any time point investigated. In this study, the effective toxic dose of DA was established at 2.5 mg/kg, as only 2 rats died after dosing (with no lesions or immunostaining) while the remaining survived the treatment and displayed clinical signs of neurotoxicity. This is a key factor in the pathogenesis of the poisoning, since only animals that exhibit neurologic signs after toxin administration develop histologic changes in the nervous system, while an overdose of DA causes death probably due to cardiac arrest. 5,17,42,53 In our experiment, at 5 to 10 minutes after the administration of DA, animals showed reduced activity and bristly hair coat, but thereafter clinical signs developed as abnormal scratching and rapidly exacerbated up to seizures. These results are in agreement with those reported by other authors in animal models inoculated with DA. 5,25,40,46,52
Studies performed on DA pharmacokinetics have demonstrated that the toxin is excreted shortly after intravenous dosing in rats, with a half-life in plasma of only 21.6 minutes 50 and complete recovery in urine within 160 minutes. 47 These data explain the lack of detection of DA by immunohistochemistry in internal organs of rats at all times after treatment in our study.
However, we observed positive staining for DA in the brain, the target organ of DA, but only at the earlier times investigated in animals dosed at 2.5 mg/kg—that is, at 6 and 10 hours after dosing and exclusively in the hippocampus. The number of immunopositive cells was greater at 6 hours than at 10 hours, a finding that suggests that at even earlier times after administration, the toxin could be detected in a greater number of cells and/or in other brain structures and, above all, that a minute quantity of DA was still present in the brain at least until 10 hours following administration. It is tempting to speculate that the presence of DA in the brain at times when the toxin is expected to be cleared from the animal may be due to the internalization of the molecule, as we detected immunoreactive DA within the cytoplasm of hippocampal pyramidal neurons, rather than extracellularly. Also, the localization of the toxin exclusively in the hippocampus after systemic administration is consistent with the idea of this brain structure as the epileptogenic center for seizures induced by the excitotoxin DA. 9,12,17
Another interesting finding in this study is that immunostaining for DA was detected only at 6 and 10 hours after treatment in the hippocampus, the major central nervous system target of the toxin. DA is a GluR agonist and is known to bind with high affinity to the AMPA/KA subclasses of ionotropic GluRs (reviewed in Jeffery et al 26 ). Although ubiquitously expressed throughout the brain, the high concentration of AMPA/KA type of receptors in the hippocampus in rodents 14,54 may account for the particular detection of DA-positive cells in this specific region of the brain. We found a more intense immunostaining for DA in CA3 hippocampal neurons at 6 hours after dosing, an area reported to contain the highest amounts of KA receptor subtype 1 in the rat brain, 54 a GluR to which DA binds with particular avidity. 26
In this study, DA administered ip did not induce histologic changes in heart, lungs, liver, and kidneys of DA-treated rats at any dose and time investigated. Lesions in these organs have not been reported to date in rats, but in the case of cynomolgus monkeys dosed intravenously with DA, some kidney or liver damage was suspected as serum biochemical parameters related with these organs appeared elevated. 40 Also, a direct toxic effect of DA on the heart has been proposed by several authors by virtue of the cardiovascular disturbances observed in humans intoxicated by DA 35,48 and because of the occurrence of myocardial lesions as a common finding in the heart of sea lions that died after a naturally occurring DA outbreak. 43 The low susceptibility of rats to DA toxicity, probably explained by the rapid renal clearance of the toxin, 47,50 may explain the absence of lesions in internal organs except the brain in this species.
DA-treated rats at a dose of 2.5 mg/kg displayed lesions in selected regions of the brain, most of them constituents of the limbic system: we found histologic changes mainly in hippocampus, amygdaloid complex, and piriform and perirhinal cortices but also in olfactory tubercle, lateral septum, and several thalamic nuclei. The preferential location of lesions in these areas of the brain has been well documented in the literature for all the species intoxicated when the dose of DA is sufficient to produce seizure activity, with only minor differences among species regarding the extent of the pathology in a particular area or the involvement of additional brain structures. 12,17,35,42,51,52
A special feature of the neurologic lesions induced by DA is that histologic changes of neuronal degeneration can be detected only by histologic observation from 24 hours onward, being most severe at 5 to 14 days after DA administration. 1 –3 We did not find lesions in the brain of rats at 24 hours following DA exposure at a dose of 2.5 mg/kg, but at 5 days, 2 distinct type of lesions were observed: selective neuronal degeneration and large areas of necrosis. The first was characterized by degenerative changes of neurons in all affected areas. This DA-induced neuronal loss found in our study is similar to that reported in the literature in the brain of animals that suffered neurologic dysfunction after exposure to DA. 12,16,42,48,51,52 In the second, large necrotic areas were predominantly found in the amygdala and piriform cortex. We are not aware of any previous observation that has reported the presence of necrosis in the brain following DA exposure in humans, rodents, or monkeys. However, a number of sea lions that died a few days after naturally occurring DA intoxication showed piriform lobe malacia on gross examination, although the authors did not report the histology of the lesion. 42 Lack of detection of extensive necrosis in previous studies may be attributed to the different histologic methods employed (HE vs Nissl staining, necrosis easily identified by using the former) and to the fact that other investigations mainly focused on the hippocampus, a structure that did not consistently show necrotic areas in the study performed on sea lions 42 and in our study. Notably, these 2 types of brain lesions (selective neuronal loss and large areas of necrosis) have been described after systemic administration of another glutamate analogue, the excitotoxin KA. 21,43 The occurrence of both types of histologic changes in specific areas of the brain is a common pathologic finding after seizure activity, which led to secondary ischemic changes and excitotoxic neurodegeneration. 12,23
At 54 day after DA exposure at a dose of 2.5 mg/kg, individual degenerating neurons were still distinguishable in damaged areas but with a different morphology than that showed at 5 days: these damaged neurons exhibited undefined borders and faintly stained perikarya and nuclei (ghost neurons). According to the literature, this gradual disintegration of damaged neurons (delayed neural death or maturation phenomenon) 23,27 induced by DA has an apoptotic component mediated by oxidative stress. 13,18 It has been reported that in the rat, hippocampal CA1 and CA3 neurons are completely absent at 3 months after DA administration. 3
Another interesting finding in this study was the presence of calcified areas associated with a strong microglial reaction in midline and dorsolateral injured thalamic nuclei at 54 days after dosing at 2.5 mg/kg. Calcium precipitation in the brain has been observed during normal aging, in association with a number of diseases, as well as after experimental inoculation of different excitotoxins. 21,32,39,44,45 Although the pathogenesis of dystrophic calcification in the nervous tissue is not fully understood, it has been considered as a strategy to reduce an excess of free intracellular Ca2+ induced by N-methyl-D-aspartate receptor activation during excitotoxicity. 10,37 As far as we know, this is the first report in the literature of selective calcification of brain lesions induced by DA.
Damaged areas in the brain of DA-treated rats contained large numbers of hypertrophied astrocytes and activated microglial cells at 5 and 54 days after dosing at 2.5 mg/kg, as revealed by GFAP immunohistochemistry and tomato lectin histochemistry, respectively. Our findings are in agreement with previous observations that reported reactive gliosis in the brain of humans and animals after intoxication with DA. 1 –3,5,45,46 In the present study, the number of microglial cells was obviously larger at 54 day compared with that seen at 5 days after DA treatment. Microglial cells were easily identified close to dying neurons, within necrotic areas and in calcified thalamic lesions, which indicates an increased phagocytic activity for debris removal and chronic neuroinflammation with release of immunologically active substances.
We also investigated the DA-induced expression of NOS in the hippocampus by using an antibody that detects the 3 enzyme isoforms (nNOS, eNOS, and iNOS). At 5 days after dosing with 2.5 mg/kg of DA, a slight increase in NOS expression was detected in pyramidal neurons and probably also in surrounding astrocytes, whereas at 54 days, immunostaining for NOS was intense and localized to cells with distribution and morphology typical of hypertrophied astrocytes. Our observations are in agreement with and further extend those previously published by Ananth et al in DA-treated rats. 1,3,4 They observed that at 5 days after dosing, hippocampal neurons expressed nNOS and astrocytes expressed iNOS; however, at 3 months after treatment, expression of nNOS was absent due to complete loss of neurons, and iNOS expression was restricted to microglial cells. Therefore, the strong immunostaining that we observed in cells with astrocyte characteristics at 54 days most probably corresponds to iNOS. It has been shown that the DA-induced excessive intracellular Ca2+ activates NOS and consequently increases the nitric oxide (NO) concentration. This overproduction of NO appears to be a major component of excitotoxic damage by several mechanisms, such as oxidative stress, DNA damage, and disruption of energy metabolism. 26,36 In fact, previous studies demonstrated that inhibition of NO production during ischemia improves survival of hippocampal pyramidal neurons by reducing or abolishing delayed neuronal death. 38 In agreement with the hypothesis proposed by Ananth et al, 3,4 the excess of production of NO by neurons may contribute to their delayed death, whereas NO produced first by astrocytes and then by microglial cells during an extended period following DA exposure may be a secondary phenomenon in the prolonged inflammatory process after neuronal injury.
Conclusions
DA systemically administered in rats at a dose of 2.5 mg/kg could be detected by immunohistochemistry in the cytoplasm of pyramidal neurons of the hippocampus at 6 and 10 hours after treatment, a finding that supports the hypothesis of this brain area as the epileptogenic center for the seizure activity induced by the excitotoxin. Animals surviving a dose of 2.5 mg/kg of DA displayed neurologic signs, and at 5 days following administration, brain damage was evident in the form of delayed death of selective neurons, extensive areas of necrosis, and a strong reactive gliosis. At 54 days after DA exposure, the pathology was characterized by still-distinguishable dying neurons, calcified lesions in the thalamus, and persistent gliosis. The expression of NOS at 5 and 54 day after dosing suggests a role for NO in the pathogenesis of neuronal degeneration and chronic inflammation induced by DA in the brain.
Footnotes
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: The research leading to these results has received funding from the following FEDER cofunded grants: CDTI and Technological Funds, supported by Ministerio de Economía y Competitividad (AGL2012-40185-CO2-01) and Consellería de Cultura, Educación e Ordenación Universitaria (GRC2013-016) and through Axencia Galega de Innovación, Spain (ITC-20133020 SINTOX, IN852A 2013/16-3 MYTIGAL). From CDTI under ISIP Programme, Spain (IDI-20130304 APTAFOOD). From the European Union’s Seventh Framework Programme managed by the Research Executive Agency (FP7/2007-2013) under grant agreements 265409 µAQUA, 315285 CIGUATOOLS, and 312184 PHARMASEA.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.