
Abstract
X-chromosome inactivation pattern (XCIP) analysis has been widely used to assess cell clonality in various types of human neoplasms. In this study, a polymerase chain reaction–based canine XCIP analysis of the androgen receptor (AR) gene was applied for the assessment of cell clonality in canine hematopoietic tumors. This XCIP analysis is based on the polymorphic CAG repeats in the AR gene and the difference of methylation status between active and inactive X chromosomes. We first examined the polymorphisms of 2 CAG tandem repeats in the AR gene in 52 male and 150 female dogs of various breeds. The 2 polymorphic CAG repeats contained 9 to 12 and 10 to 14 CAGs in the first and second CAG repeats, respectively. Of the 150 female dogs, 74 (49.3%) were heterozygous for the first and/or second polymorphic CAG tandem repeats, indicating the utility of XCIP analysis in these dogs. Canine XCIP analysis was then applied to clinical samples from female dogs with canine high-grade lymphoma, chronic myelogenous leukemia, acute myelogenous leukemia, and benign lymph node hyperplasia. Of 10 lymphoma cell samples, 9 (90%) showed skewed XCIPs, indicating their clonal origins, whereas all the nonneoplastic lymph node samples showed balanced XCIPs. Moreover, bone marrow specimen from a dog with acute myelogenous leukemia and peripheral leukocyte specimens from 2 dogs with chronic myelogenous leukemia showed skewed XCIPs. XCIP analysis was successfully employed to demonstrate the cell clonality of canine hematopoietic tumors in this study and will be applicable to evaluate the clonality in various proliferative disorders in dogs.
Clonal cell proliferation is a fundamental characteristic of neoplasms. Neoplastic cells are essentially derived from a single transformed cell, whereas normal or reactive tissues are generally composed of polyclonal cell populations. Assessment of clonality can provide useful information for diagnosis, management, and understanding of the pathogenesis of proliferative disorders.
In human medicine, several analytic systems have been developed for assessment of cell clonality. They include the assessments of aberrant phenotype, specific chromosome/gene abnormalities, antigen receptor gene rearrangement, or X-chromosome inactivation pattern (XCIP). 9,11 Each method can be applied to certain kinds of neoplastic proliferative diseases, but each also has its limitations.
Of the analytic methods used to assess clonality in humans, XCIP analysis was chosen for this study because it can be applied to any type of neoplastic diseases, even those in which no tumor-specific phenotype or genetic abnormality has been identified. In mammalian females, either the paternal or maternal X chromosome generally undergoes random inactivation in each cell through the epigenetic changes during embryogenesis. This event is stably inherited by the daughter progeny of each cell, resulting in the theoretical expectation of an approximately 1:1 inactivation ratio between the paternally and maternally derived X chromosomes in normal or reactive tissues. By contrast, most neoplastic tissues are composed of a population clonally expanded from a single cell, generally exhibiting a uniform XCIP and resulting in its significant deviation from 1:1 inactivation ratio. Therefore, demonstration of a nonrandom XCIP (skewed XCIP) is indicative of the clonal origin of cells. 9
XCIP analysis has been widely used for the assessment of cell clonality in various neoplastic and preneoplastic conditions in humans, such as chronic myelogenous leukemia (CML), 12 acute myelogenous leukemia (AML), 35 myelodysplastic syndrome, 8 lymphoma, 27 and certain nonhematopoietic tumors. 18,27,36 Although XCIP analysis is applicable only in female patients, a major advantage of this analysis is its possible application to many types of neoplastic diseases, even those in which a tumor-specific marker has not been identified.
A widely used method in humans for determining clonality by using the X-chromosome inactivation principle is the human androgen receptor (AR) gene assay (HUMARA). Transcription of the human AR gene on the inactive X chromosome is strictly inhibited by methylation of the CpG island in the promoter. 17 A similar epigenetic event is known to occur on a region containing the methylation-sensitive endonuclease HpaII sites near the polymorphic CAG tandem repeat (CAGr) in exon 1 of the AR gene. 1 Because of the difference in methylation status between inactive and active X chromosomes, DNA from only inactive (methylated) X chromosomes can be amplified after HpaII digestion, through polymerase chain reaction (PCR) with primers flanking the restriction sites. A difference in the number of the CAG repeats between 2 X chromosomes enables their distinction from each other.
Similar to the human AR gene, the canine AR gene is located on the X chromosome, and it contains 2 clusters of polymorphic CAGr in its exon 1 (CAG-1 and CAG-2 in Fig. 1; GenBank accession No. NC_006621.3). By using these polymorphic CAGrs, researchers developed canine XCIP analyses. 2,33 The clonality was recently demonstrated in canine histiocytoma via this principle. 10 In this study, we evaluated the clonality of canine hematopoietic tumors using XCIP analysis, and we demonstrated clonal XCIPs in lymphoma and leukemia cells.
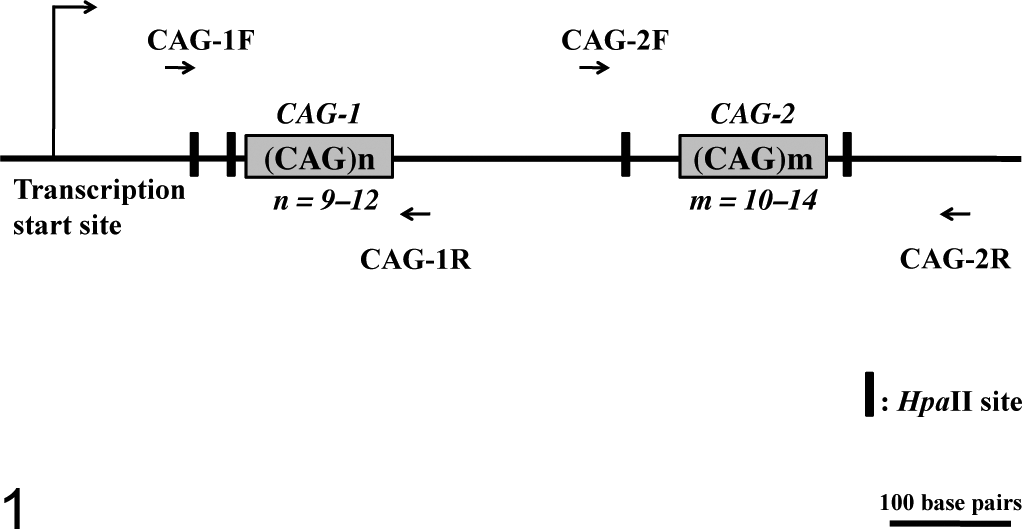
Schematic structure of the canine AR gene exon 1, based on the registered sequences of the canine AR gene (GenBank accession No. NC_006621.3). Two polymorphic CAG tandem repeats (CAG-1 and CAG-2) are located in exon 1 of the canine AR gene. The polymorphic CAG-1 and CAG-2 loci can be amplified by PCR through CAG-1F and CAG-1R primers and CAG-2F and CAG-2R primers, respectively. Two HpaII restriction sites within the sequences are amplified by CAG-1F and CAG-1R and by CAG-2F and CAG-2R.
Methods
Samples for Characterization of the AR Gene in Dogs
Peripheral blood samples were obtained from 52 male and 150 female dogs; they included 12 healthy blood donors and 190 patients referred to the Veterinary Medical Center of the University of Tokyo for the diagnosis and treatment of various diseases unrelated to hematopoietic neoplasms. Use of the blood samples from the dogs for research purposes was approved by the owners in written statements. The breeds of the 202 dogs were Miniature Dachshund (n = 31), mixed (13), Shih Tzu (13), Chihuahua (10), Maltese (10), Pembroke Welsh Corgi (10), Beagle (9), Labrador Retriever (9), Yorkshire Terrier (9), American Cocker Spaniel (7), Miniature Schnauzer (7), Pomeranian (7), Cavalier King Charles Spaniel (6), French Bulldog (6), Golden Retriever (6), Pug (6), Toy Poodle (6), Shetland Sheepdog (5), West Highland White Terrier (5), Shiba (4), Bernese Mountain Dog (2), English Springer Spaniel (2), Italian Greyhound (2), Miniature Pinscher (2), and 15 other purebred dogs (1 each).
Clinical Samples for XCIP Analysis
The samples used were cells of hematopoietic tumors (n = 25 samples) or nonneoplastic lymph nodes (n = 19) obtained from female dogs referred to the Veterinary Medical Center of the University of Tokyo between 2007 and 2011. The ages of the 25 dogs with hematopoietic tumors ranged from 10 months to 11 years (median, 6 years). Fifteen dogs were spayed and 10 were intact. The breeds were Golden Retriever (n = 3), Miniature Dachshund (3), Pembroke Welsh Corgi (3), French Bulldog (2), Labrador Retriever (2), Miniature Schnauzer (2), Shih Tzu (2), mixed (1), and 7 other purebred dogs (1 each).
The 25 hematopoietic tumor samples comprised lymph node samples of multicentric high-grade lymphoma according to Kiel classification 13 (n = 21 dogs), bone marrow samples of AML (2; AML-M5 and AML-M6 according to FAB classification 16 ), and peripheral blood samples of CML (2). The diagnoses of these neoplasms were made by cytologic analysis. CML was diagnosed in dogs with persistent (> 1 month) and marked leukocytosis (> 50 000/μl of peripheral leukocytes) by exclusion of other possible reactive leukocytosis. 15
Before the XCIP analysis, we evaluated clonality in lymphoma samples by analyzing rearrangement patterns of immunoglobulin heavy-chain and T-cell receptor γ genes (IgH and TCRγ, respectively), using PCR as described before. 5 Of the 21 lymphoma cases, 20 were shown to have clonal rearrangement of IgH gene, whereas the remaining 1 case had clonal TCRγ gene rearrangement. The immunophenotypes of the neoplastic cells of AML-M5 and AML-M6 were assessed by flow cytometry via primary monoclonal antibodies against CD3 (CA17.2A12, Leukocyte Antigen Biology Laboratory, Davis, CA), CD4 (YKIX 302.9, AbD Serotec, Oxford, UK), CD8 (YCATE 55.9, AbD Serotec), CD14 (TUK4, Santa Cruz Biotechnology Inc, Santa Cruz, CA), CD18 (CA1.4E9, Leukocyte Antigen Biology Laboratory), CD21 (CA2.1D6, AbD Serotec), CD34 (1H6, BD Bioscience, San Diego, CA), CD45 (CA12.10C12, Leukocyte Antigen Biology Laboratory), and MHC class II (CA2.12C12, Leukocyte Antigen Biology Laboratory) and a secondary antibody against mouse IgG1 (BD Bioscience) as described previously. 24 Neoplastic cells from a dog with AML-M5 stained positive for CD45, CD14, and CD18 and were negative for CD3, CD4, CD8, CD21, CD34, and MHC class II; thus, these cells were shown to be monocytic and therefore of monocytic lineage. Neoplastic cells from a dog with AML-M6 were negative for all the antigens described above.
Nineteen nonneoplastic lymph node samples were obtained from dogs with lymphadenopathy due to nonneoplastic conditions such as pyoderma and demodicosis. The polyclonal origin of the lymphocyte population was determined on the basis of cytologic examination and the absence of the clonal antigen receptor gene rearrangement.
DNA Extraction
Genomic DNA samples were isolated from cells by DNeasy Blood and Tissue Mini Kit (QIAGEN, Hilden, Germany). Concentration of the extracted genomic DNA samples was calculated on the basis of absorbance at 260 nm, as measured with a spectrophotometer.
Sequencing of CAGr in the Canine AR Gene in Male Dogs
Primer pairs were designed per the sequence of the canine AR gene (NC_006621.3) by Primer3Plus (http://www.bioinformatics.nl/cgi-bin/primer3plus/primer3plus.cgi) for amplification of a DNA fragment spanning the first (CAG-1, NC_006621.3; nucleotide [nt] 163–192) and second (CAG-2, nt 538–570) CAGr in its exon 1 (AR-F, nt 45–63, 5′–GTCCAAGACCTATCGAGGA–3′ and AR-R, nt 694–713, 5′– CTGTCCGAGATGGTCGAACT–3′). Each reaction mixture contained 50 ng of genomic DNA, 2.0 mM MgCl2, 0.2 mM of each dNTP, 0.5 μM of each primer, 1× PCR buffer, and 2 units of ExTaq DNA polymerase (Takara Bio Inc, Otsu, Japan) in a total volume of 20 μl. Cycle conditions consisted of an initial denaturation step at 95°C for 1 minute, followed by 35 cycles of denaturation at 94°C for 30 seconds, annealing at 60°C for 30 seconds, and extension at 72°C for 1 minute, with a final extension at 72°C for 7 minutes. PCR products were purified with a DNA purification kit (SUPREC PCR, Takara Bio Inc, Otsu, Japan). Sequence analysis of the PCR products was carried out with BigDye Terminator v. 3.1 Cycle Sequencing Kit (Applied Biosystems, Foster City, CA) and the Genetic Analyzer 3130xl (Applied Biosystems) through the primers AR-F and AR-R. The repeat number of the 2 CAGrs in each dog was counted with Sequence Scanner Software v. 1.0 (Applied Biosystems).
Examination of the Heterozygosity of CAG-1 and CAG-2 in the Canine AR Gene in Female Dogs
DNA fragments containing CAG-1 or CAG-2 were amplified by PCR with the primers reported previously (for amplification of CAG-1: CAG-1F primer, NC_006621.3, nt 93–112, 5′–CGAAGTGATCCAGAACCCGG–3′ and CAG-1R primer, nt 294–272, 5′–TTCCTCATCCAGAGCCAGGTAGC–3′; for amplification of CAG-2: CAG-2F primer, nt 435–456, 5′–CCCATCCACATTGTCACTGCTG–3′ and CAG-2R primer: nt 750–730, 5′–CATGGACACCGACACTGCCTT–3′). 2 The forward primers were labeled with 6-FAM for capillary electrophoresis. Each reaction mixture contained 50 ng of genomic DNA, 2.0 mM MgCl2, 0.2 mM of each dNTP, 0.5 μM of each primer, 1× PCR buffer, and 2 units of ExTaq DNA polymerase (Takara Bio Inc, Otsu, Japan) in a total volume of 20 μl.
Cycle conditions consisted of an initial denaturation step at 95°C for 1 minute, followed by 28 cycles of denaturation at 95°C for 30 seconds, annealing at 60°C for 30 seconds, and extension at 72°C for 30 seconds, with and a final extension at 72°C for 7 minutes. One microliter of the 10-fold diluted PCR product was mixed with 8.5 μl of formamide and 0.5 μl of 600LIZ size standard (Applied Biosystems) in a 96-well plate. These samples were denatured at 95°C for 5 minutes and immediately placed on ice for 15 minutes. The length of the PCR product was determined by capillary electrophoresis using the Genetic Analyzer 3130xl and evaluated by GeneMapper software (Applied Biosystems).
XCIP Analysis With the AR Gene
In this study, we utilized the XCIP analysis method reported by Bell et al, 2 with some modifications. DNA samples (500 ng) were incubated with 5 units of a methylation-sensitive endonuclease, HpaII (New England Biolabs Inc, Ipswich, MA), in 1× buffer 1 in a total volume of 50 μl. Undigested controls of each sample were incubated in 1× buffer1 (total volume, 50 μl) without HpaII. Both the digested and undigested samples were incubated for 16 hours at 37°C, followed by inactivation for 20 minutes at 65°C. The same amount of DNA from a male nonneoplastic peripheral blood sample was used as a control for HpaII digestion because male somatic cells contain only 1 unmethylated (active) X chromosome.
A 50-ng aliquot of the digested and undigested DNA samples was subjected to subsequent PCR under the same PCR conditions and with the same primers described above (CAG-1F and CAG-1R for amplification of CAG-1 locus and/or CAG-2F and CAG-2R for amplification of CAG-2 locus). These amplified regions each contain 2 HpaII sites within the region (Fig. 1).
The results of XCIP analysis were evaluated after confirming the absence of PCR amplification from the completely digested male somatic DNA control. The degree of skewness (imbalanced inactivation) was determined by comparing the area under the curve (AUC) for each allele of the digested and undigested samples via the results of the capillary electrophoresis. The corrected inactive allele ratio (CIAR) was defined as the allele ratio of the digested sample divided by the allele ratio of the undigested sample, and it was determined to avoid the influence of potential preferential amplification of either allele.
Stutter peaks (shadow bands) generated by slippage synthesis of short tandem repeats are observed as minor peaks 3 base pairs shorter than real peaks.
34
When the CAG tandem repeat in one allele is longer than that in another allele by 3 base pairs (1 CAG repeat), the longer allele affects the measured AUC of the shorter allele. To avoid the influence of shadow bands, the percentage of shadow bands for each repeat length was calculated from the data obtained from DNA samples of male dogs. If the sizes of 2 alleles at CAG-1 or CAG-2 differed by 1-repeat length, 15% or 20% of the AUC of longer allele was subtracted from that of shorter allele, respectively. Thus, CIAR was calculated as follows:
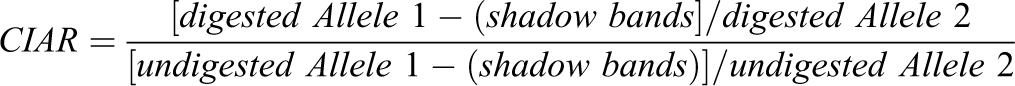
As proposed in previous reports on XCIP analysis in humans, 20,22 the sample population was judged to be clonal when CIAR > 3.0. For ease of comparison, where CIAR < 1, the corrected ratio was calculated as the inverse value of the CIAR. If dogs were heterozygous for both CAG-1 and CAG-2, the higher CIAR was used for analysis. All reactions were run in duplicate to ensure that the results were reproducible.
Statistical Analysis
Ages between dogs with hematopoietic tumors and those with nonneoplastic lymph nodes were compared with the Wilcoxon rank-sum test via statistical software R. 28 Significance was set at P < .05.
Results
Polymorphism of Repeat Number at CAG-1 and CAG-2 in Male and Female Dogs
The number of CAG trinucleotide repeats in the CAG-1 and CAG-2 loci was examined in 52 male dogs. The number was 10 or 11 at CAG-1, and it varied between 11 and 14 at CAG-2. The absence of single nucleotide polymorphisms was confirmed at the 4 HpaII sites used for the XCIP analysis.
Next, we performed PCR amplification of CAG-1 and CAG-2, followed by capillary electrophoresis for DNA samples from 150 female dogs. In homozygotes for these CAGrs, a single major peak was observed (Fig. 2a, d). Heterozygotes were identified by the occurrence of 2 major peaks of different lengths after PCR amplification encompassing CAG-1 or CAG-2 (Fig. 2b, c, e, f). In all the samples, stutter peaks (shadow bands indicated as S) were observed as minor peaks, shorter than the major peaks by 3 base pairs (Fig. 2).
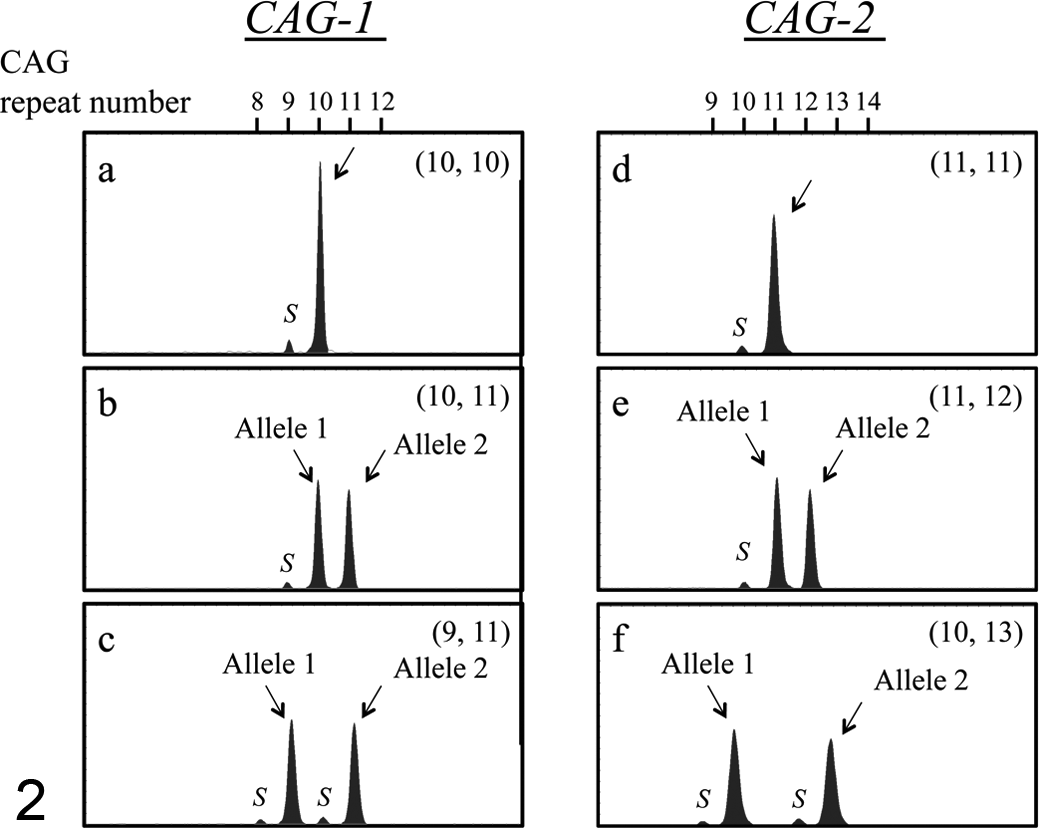
Representative capillary electrophoretograms after PCR to amplify the polymorphic CAG tandem repeats, CAG-1 (a–c) and CAG-2 (d–f), in the canine AR gene (a, d). Nonneoplastic blood DNA samples from female dogs homozygous for the repeat number of CAG trinucleotides (a: 10 CAG repeats at CAG-1; d: 11 CAG repeats at CAG-2). (b, c, e, f) Nonneoplastic blood DNA samples from female dogs heterozygous for the repeat number of CAG trinucleotides (b: 10 and 11 CAG repeats at CAG-1; c: 9 and 11 CAG repeats at CAG-1; e: 11 and 12 CAG repeats at CAG-2; f: 10 and 13 CAG repeats at CAG-2). The numbers in parentheses are the numbers of CAG repeats of alleles 1 and 2. Stutter peaks (shadow bands, indicated as S) are observed as minor peaks shorter than the major peaks by 3 base pairs.
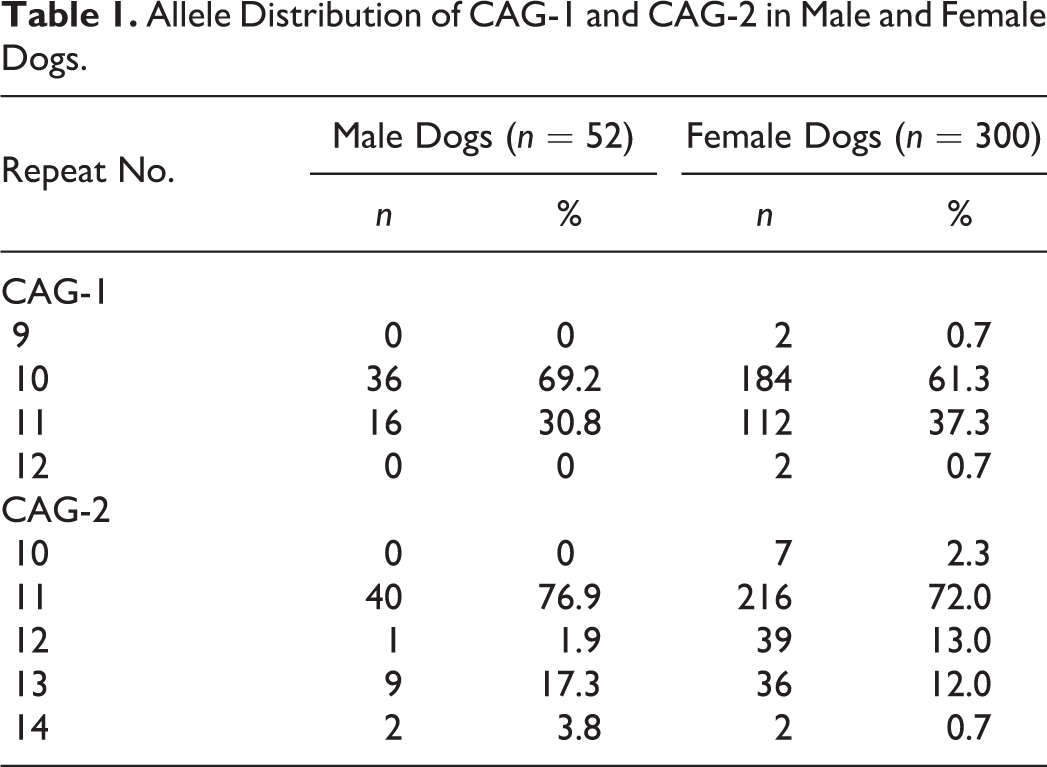
Of the 150 female dogs, 47 (31.3%) and 51 (34%) were found to be heterozygous for CAG-1 and CAG-2, respectively. Heterozygosity of at least 1 locus was shown in 74 of 150 (49.3%) female dogs, indicating the possible usefulness of XCIP analysis in these dogs. The allele frequency with respect to the number of CAG repeat in our study population is summarized in Table 1.
Allele Distribution of CAG-1 and CAG-2 in Male and Female Dogs.
XCIP Analysis of Clinical Samples
To examine the applicability of XCIP analysis in clinical samples, 25 neoplastic and 19 nonneoplastic samples were examined for the heterozygosity of CAG-1 and CAG-2 before XCIP analysis. Of the 25 neoplastic cell DNA samples, 13 samples (high-grade lymphoma, 10 dogs; CML, 2 dogs; and AML-M5, 1 dog) were heterozygous for CAG-1 and/or CAG-2 and were therefore appropriate for XCIP analysis. Similarly, of the 19 DNA samples from nonneoplastic lymph nodes, 12 (63%) were heterozygous for CAG-1 and/or CAG-2. The DNA samples from dogs heterozygous for CAG-1 and/or CAG-2 were used for further XCIP analysis. Signalment and the AR gene polymorphism of dogs analyzed by XCIP analysis are summarized in Table 2. There was no statistical difference in ages between the dogs with hematopoietic tumors and those with nonneoplastic lymph nodes.
Signalment and the AR Gene Polymorphism of Dogs Analyzed by XCIP Analysis.a
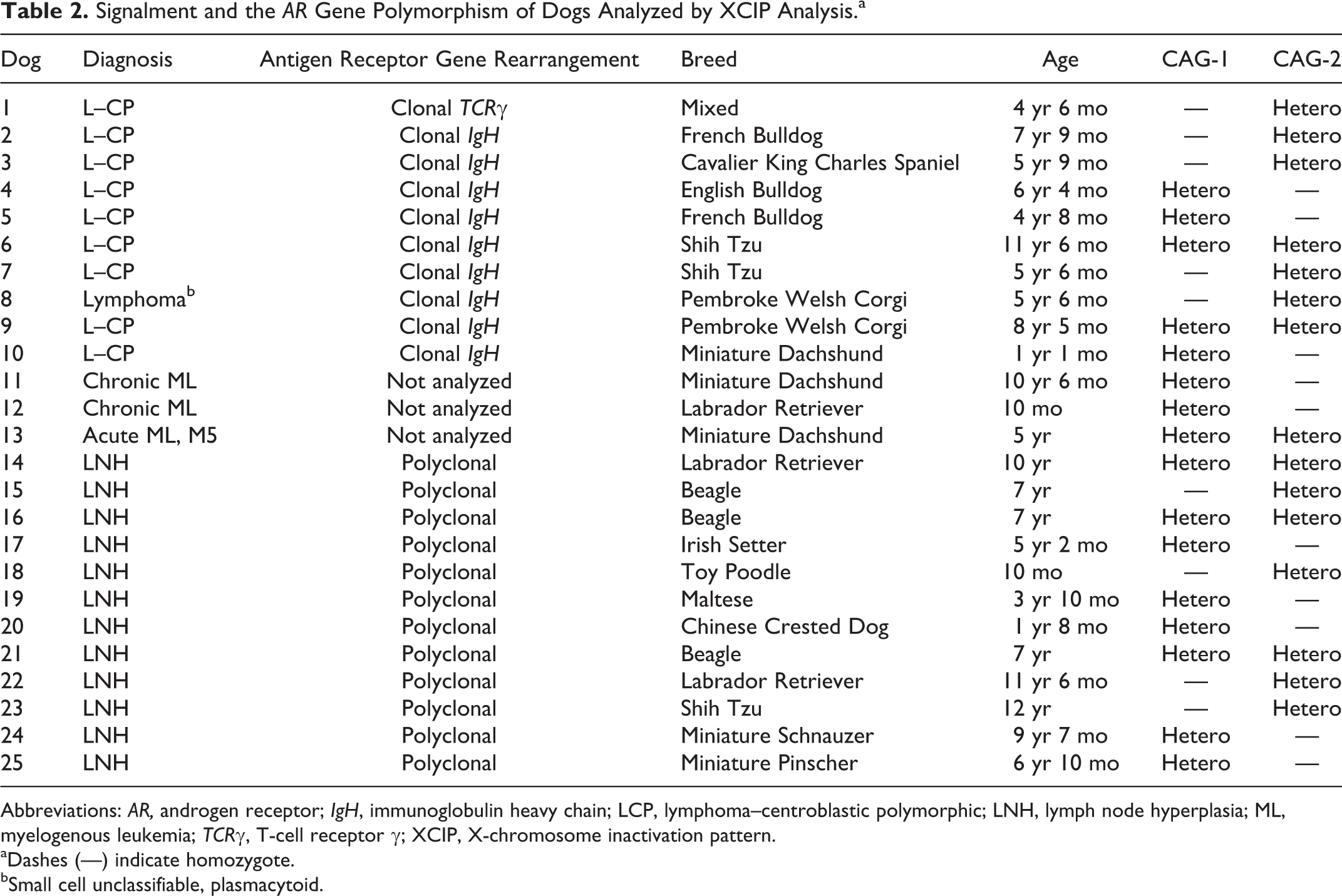
Abbreviations: AR, androgen receptor; IgH, immunoglobulin heavy chain; LCP, lymphoma–centroblastic polymorphic; LNH, lymph node hyperplasia; ML, myelogenous leukemia; TCRγ, T-cell receptor γ; XCIP, X-chromosome inactivation pattern.
aDashes (—) indicate homozygote.
bSmall cell unclassifiable, plasmacytoid.
Of the 10 samples of high-grade lymphoma cells, CIARs in 9 samples exceeded 3.0 (3.26–∞), indicating skewed XCIPs (Fig. 3c–e); however, 1 DNA sample showed balanced XCIP (CIAR: 1.13). The CIAR of 1 sample was calculated as ∞ because HpaII digestion completely inhibited PCR amplification of active allele, causing the AUC of either allele to approach 0 (Fig. 3e). However, all nonneoplastic DNA samples obtained from nonneoplastic lymph nodes showed balanced XCIPs (CIARs: 1.02–2.67; median: 1.63) (Fig. 3b).
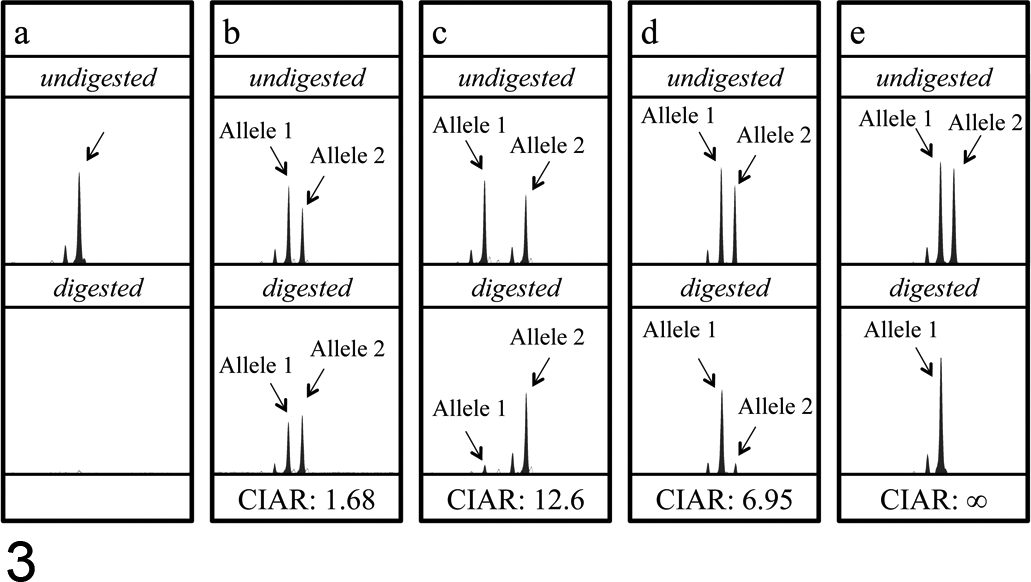
Representative results of XCIP analysis in nonneoplastic and neoplastic cell samples. (a) A nonneoplastic peripheral blood cell sample from a male dog. A peak corresponding to the PCR-amplified CAG-2 is observed in an undigested sample but not in the HpaII-digested sample, indicating complete digestion with HpaII. (b) A nonneoplastic lymph node cell sample from a female dog heterozygous for CAG-2. Because the cells are polyclonal, the AR gene fragments on alleles 1 and 2 were almost equally amplified in both the undigested and HpaII-digested samples (CIAR: 1.68). (c, d, e) Canine lymphoma cell samples (c: case 1; d: case 6; e: case 10). The CAGrs (CAG-2 in cases 1 and 6 and CAG-1 in case 10) were similarly amplified in the undigested samples; however, the amount of PCR product from 1 allele (allele 1 in c, allele 2 in d and e) was markedly reduced in the HpaII-digested samples. The CIARs of these samples were 12.6, 6.95, and ∞, exceeding the threshold value 3.0, indicating clonal cell proliferation in these samples. Stutter peaks are observed as peaks 3 base pairs shorter than the major peaks in all the electrophoretograms.
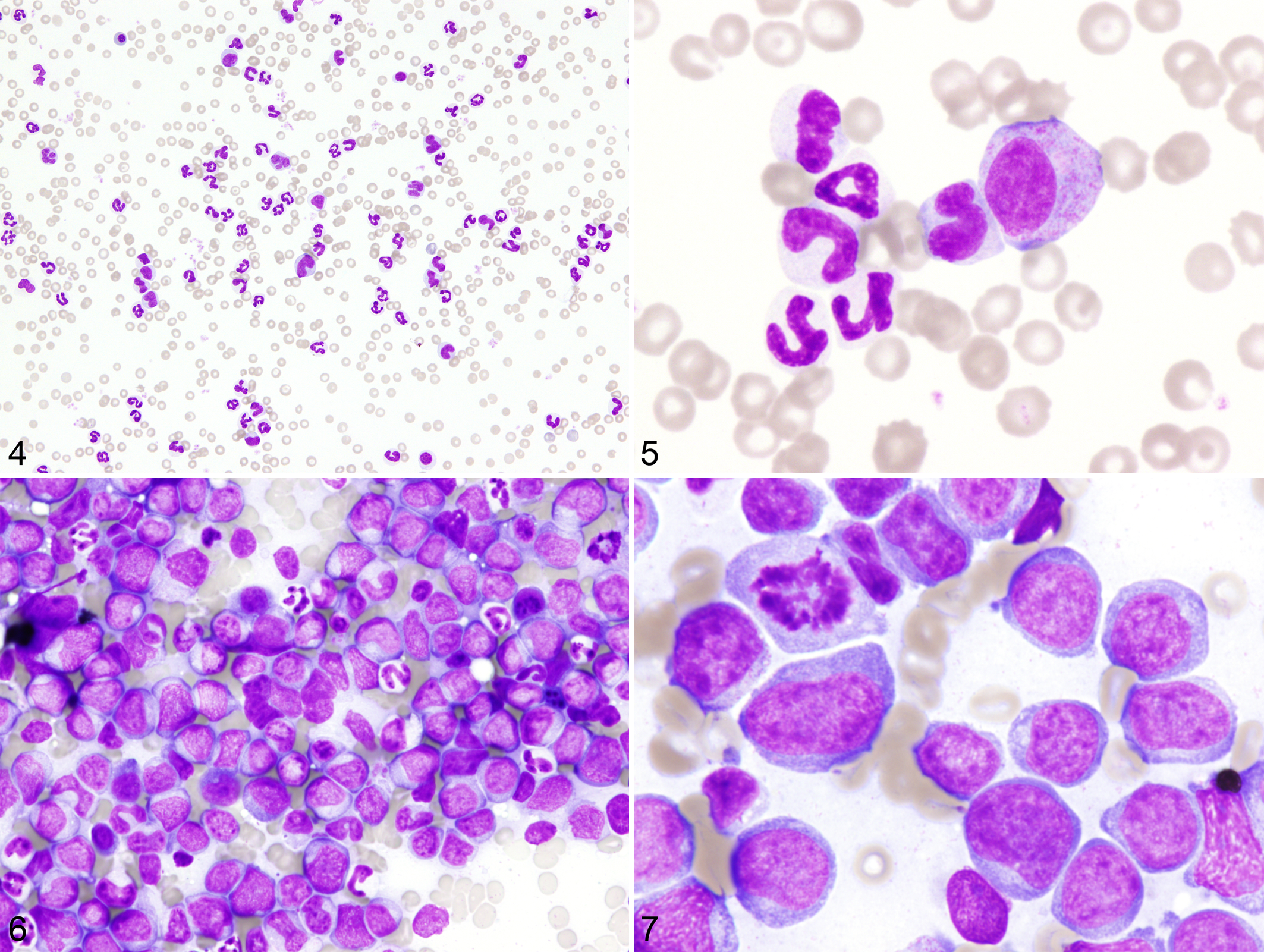
Marked leukocytosis predominantly composed of mature and immature granulocytes in the absence of inflammatory lesions in the body was a characteristic feature in the peripheral blood of the 2 dogs with CML (Figs. 4, 5). Bone marrow specimens of these dogs were hypercellular with increase in the granulocytic cell lineage, resulting in high myeloid–erythroid cell ratio, and they lacked apparent dysplastic features. In the dog with AML-M5, more than 90% of the bone marrow cells were neoplastic monocytes. Normal hematopoiesis was partially replaced by tumor cell infiltration (Figs. 6, 7). All 3 samples from dogs with CML or AML-M5 showed extremely skewed XCIPs (Fig. 8). The CIARs of neoplastic and nonneoplastic samples used in the present study are summarized in Figure 9.
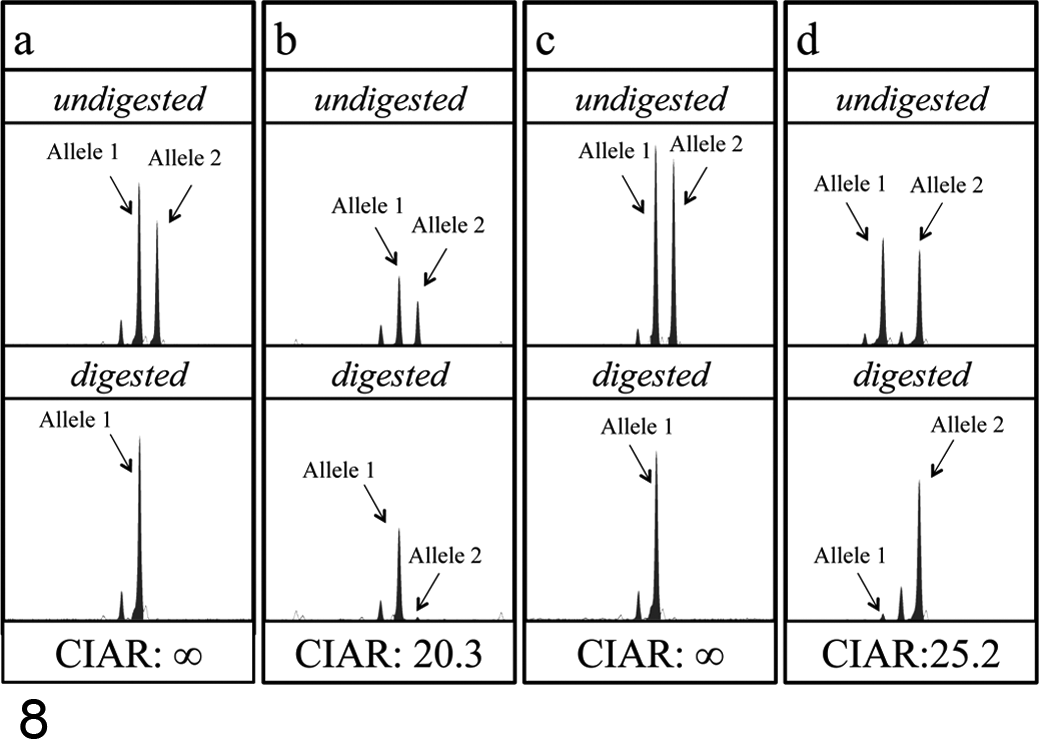
XCIP analysis in neoplastic cells obtained from dogs with CML or AML. (a, b) Peripheral blood samples from dogs with CML (a: case 11; b: case 12). DNA fragments derived from alleles 1 and 2 containing CAG-1 on both alleles were similarly amplified in the undigested samples; however, the amount of the PCR product from the allele 2 was markedly reduced in the HpaII-digested samples in both cases. (c, d) A bone marrow sample from a dog with AML-M5 (case 13) (c: XCIP analysis at CAG-1; d: XCIP analysis at CAG-2). DNA fragments containing CAG-1 or CAG-2 on both alleles were similarly amplified in the undigested samples; however, the amount of PCR product from one allele (allele 2 in c, allele 1 in d) was markedly reduced in the HpaII-digested samples.
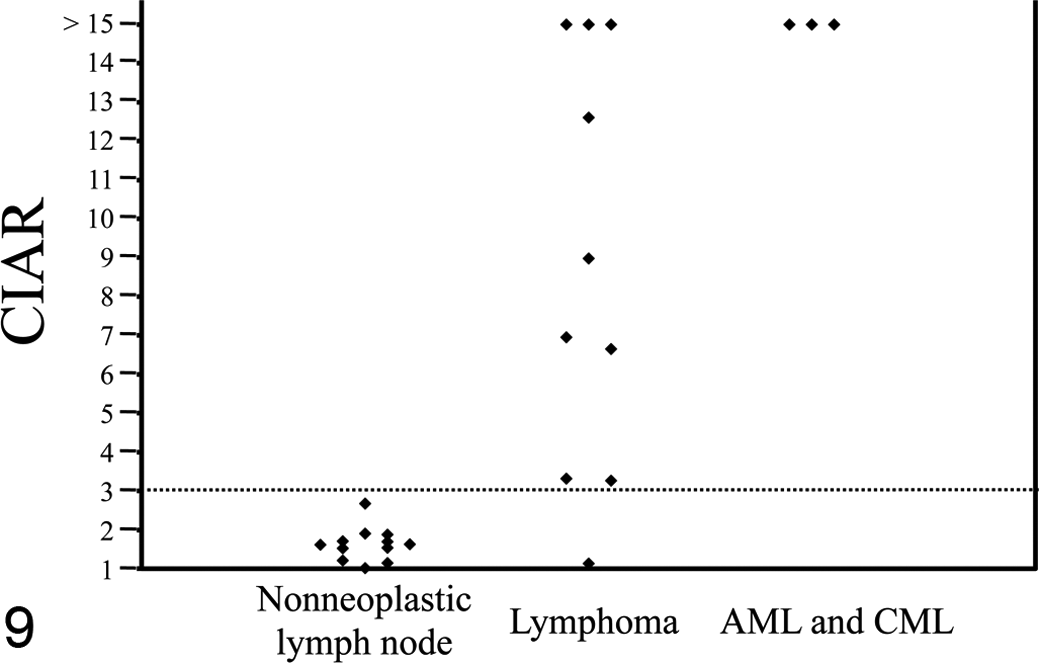
CIAR obtained from the XCIP analysis of 12 nonneoplastic lymph node samples, 10 lymphoma samples, and 3 myeloid leukemia samples. The threshold value of CIAR was set at 3.0.
Of the 6 samples heterozygous for both CAG-1 and CAG-2, the clonality status judged by CIAR agreed between CAG-1 and CAG-2 in 4 samples but disagreed in 2 lymphoma samples (CIARs < 3.0 at CAG-1 and CIARs >3.0 at CAG-2).
Discussion
In the present study, we first examined the frequency of the heterozygosity of polymorphic CAGrs in the AR gene of 150 female dogs of various breeds to know the applicability of XCIP analysis of AR gene in dogs. We then applied canine XCIP analysis for the evaluation of cell clonality in clinical tumor samples of hematopoietic origins.
In the study reported here, approximately 50% of the female dogs were heterozygous for CAG-1 and/or CAG-2. This result is consistent with that of a previous study, where 41% of 27 female dogs were heterozygous for CAG-1 and/or CAG-2. 29 However, another study showed only 19% of 131 female dogs were heterozygous. 10 This marked difference may reflect the difference in the genetic diversity among the dog populations analyzed in these studies. The AR genes of other mammals, such as humans and cats, have been reported to contain 1 polymorphic CAGr with higher frequency of heterozygosity (90% in human female individuals 1,14 and 68% in female cats 23 ). The relatively low frequency of the heterozygosity in the canine AR gene may be due to the low genetic diversity within purebred dogs as a result of selective breeding after domestication.
Skewed XCIPs were found in 9 of the 10 lymphoma samples examined in this study, indicating clonal cell populations in these cells, which was consistent with the cytologic findings and the results of antigen receptor gene rearrangement analysis. However, 1 lymphoma sample did not show skewed XCIP. There are several possible explanations for this result. First, because the present assay is based on differences in the methylation status between 2 X chromosomes, the CIAR would not reflect the actual ratio of the inactivation pattern of the 2 X chromosomes when abnormal hypermethylation or hypomethylation occurs at the AR locus. Abnormal hypermethylation of the AR locus has been reported in human lymphoma patients. 21 Moreover, CIARs were found to disagree between CAG-1 and CAG-2 in the 2 lymphoma samples analyzed in this study, which could probably be attributable to the difference in the methylation pattern at the HpaII sites in the CAG-1 and CAG-2 loci. Recently, incomplete correlation was also reported with DNA methylation pattern at the human AR gene and X-chromosome inactivation. 31 The incomplete methylation may lead to this disparity in clonality status assessed by 2 loci. Therefore, to avoid false polyclonal patterns, more than 1 locus should be analyzed. Second, successful detection of unbalanced XCIPs largely depends on the proportion of tumor cells in the samples used for the assay. If the samples contained a large number of nonneoplastic cells (theoretically, > 50%), false-negative results would be obtained. However, because microscopic analysis showed that neoplastic cells constituted more than 80% of the cells in the neoplastic specimens, occurrence of false-negative results seems unlikely.
Demonstration of cell clonality is difficult in proliferative diseases except for those of lymphoid cell lineage. The lack of a clonality analysis has been especially problematic in the diagnosis of various types of chronic myeloproliferative neoplasms (CMPNs), 19 which are a group of diseases characterized by overproduction of bone marrow cells and considered to arise from clonal expansion of an abnormal hematopoietic stem cell. This class of disease includes polycythemia vera, essential thrombocythemia, and CML. Proliferating cells in CMPNs differ little from normal blood cells in their morphology, making discrimination between neoplastic proliferating cells and normal/reactive proliferating cells difficult. Diagnosis of CMPNs can be made only by exclusion of other causes after thorough clinical examination; however, it is sometimes impossible to obtain a definitive diagnosis. As is the case in other CMPNs, CML can be diagnosed only by exclusion of other causes of reactive leukocytosis because the proliferating cells show little apparent morphologic abnormalities. However, a recurrent chromosomal translocation has been recently identified in dogs with CML, implying their neoplastic proliferation. 4 In the present study, clonality of the proliferating leukocytes in dogs with clinical and hematologic diagnosis of CML was also confirmed on the basis of skewed XCIPs. Further studies on the molecular biological and cytogenetic characterization of canine CMPNs would provide information that can be used for improving the diagnosis and treatment of various CMPNs in dogs.
In addition to CMPNs, clonality of proliferating cells is still unclear in a variety of other diseases in dogs, including splenic nodular hyperplasia/fibrohistiocytic nodules, 30 myelodysplastic syndromes, 3,15 and histiocytic proliferative diseases, 25 making the management of these diseases difficult. XCIP analysis could be used to assess the clonality status, which would help design better strategies for the management of these diseases.
There are 2 limitations in this study that we need to address. First, small numbers of neoplastic samples did not allow us to analyze neoplastic samples extensively by additional parameters, such as tumor types and the degree of XCIP skewness, although the difference in XCIPs between neoplastic and nonneoplastic samples was clearly demonstrated. Second, our lymphoma cases were classified by the classical Kiel classification, not by the current World Health Organization classification system, 32 due to the unavailability of histopathologic examination and immunohistochemical examination, making it difficult to compare the result with those of future studies. Given the fact that most of our lymphoma cases were centroblastic polymorphic type and showed clonal IgH gene rearrangement, most lymphoma cases were likely to be classified as diffuse large B-cell lymphoma in the World Health Organization classification.
Although we believe that XCIP analysis can be a useful tool to evaluate cell clonality, recent studies suggest that there may be a potential pitfall in using XCIPs as a clonality analysis, especially for hematopoietic diseases. 6,26 In these studies, clonal hematopoiesis was demonstrated in peripheral leukocytes in elderly women. In addition, the proportion of genes escaping inactivation process differs among species and among individuals within the same species. 7 Although skewed XCIPs were not observed in any samples of nonneoplastic lymph nodes in this study, as well as in 3 samples of mammary glandular hyperplasia in a previous study, 10 we must keep in mind that this age-related skewness and the variability of X chromosome inactivation may also occur in female dogs. Further research is warranted to evaluate XCIPs in normal cells as well as those in neoplastic ones.
Another limitation of XCIP analysis is that the current assay cannot be applied to dogs that are homozygous for both CAG-1 and CAG-2, approximately 50% of the female dogs. Availability of this assay depends on the polymorphism of the 2 CAGrs. Therefore, to apply XCIP analysis to a wider female population, assays based on other X-linked polymorphic genes need to be developed.
Footnotes
Acknowledgements
We thank Drs Kunio Shiota and Jun Ohgane for their helpful discussions.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This study was supported by the Japan Society for the Promotion of Science (KAKENHI 23381082).