
Abstract
Veterinary pathology of infectious, particularly viral, and neoplastic diseases has advanced significantly with the advent of newer molecular methodologies that can detect nucleic acid of infectious agents within microscopic lesions, differentiate neoplastic from nonneoplastic cells, or determine the suitability of a targeted therapy by detecting specific mutations in certain cancers. Polymerase chain reaction–based amplification of DNA or RNA and in situ hybridization are currently the most commonly used methods for nucleic acid detection. In contrast, the main methodology used for protein detection within microscopic lesions is immunohistochemistry. Other methods that allow for analysis of nucleic acids within a particular cell type or individual cells, such as laser capture microdissection, are also available in some laboratories. This review gives an overview of the factors that influence the accurate analysis of nucleic acids in formalin-fixed tissues, as well as of different approaches to detect such targets.
Technical Considerations
Tissue Handling, Fixation, and Embedding
Fixation of tissues in 10% neutral buffered formalin, followed by paraffin embedding (FFPE), has long been the standard approach of tissue preparation for microscopic evaluation. The chemical principle of formalin fixation is based on the cross-linking of proteins. Formalin has been used extensively because it is inexpensive and excellently preserves morphological detail in histological sections. However, the relatively recent development and increasing use of both protein and nucleic acid detection methods have revealed some significant shortcomings associated with the formalin-based fixation process. The effects of formalin fixation on DNA are chemical modification, DNA trapping, and potentially DNA fragmentation. Chemical modification, considered the main effect, results from the direct cross-linking of proteins to DNA. 68 In addition, extensive protein-protein cross-linking and the formation of protein networks result in DNA trapping. The combination of these effects makes it much more difficult to extract DNA from FFPE tissue than from unfixed tissue, especially when methods used for extraction from fresh tissues are used on FFPE tissue without any modifications. 60,83
In addition to interfering with the ability to extract nucleic acid, formalin fixation also causes DNA fragmentation, especially when the formalin used is inadequately buffered, which then leads to formic acid formation during the fixation process. This, in turn, results in depurination of the DNA, which then becomes susceptible to cleavage by hydroxyl ions and results in the formation of single-stranded breaks. 60 Similar to DNA, RNA is also degraded and chemically modified as the result of formalin fixation. The most important chemical effect of formalin on RNA is the formation of monomethylol groups with purine bases. 77 This has a significant effect on reverse transcription when reverse transcription polymerase chain reaction (RT-PCR) is the downstream molecular assay used. Damage to messenger RNAs (mRNAs) also specifically results from modification of polyA tails. 79 Fragmentation of RNA is another potential problem and mainly the result of ubiquitous RNAse activity. This effect is primarily the result of a prolonged time interval between tissue collection and fixation, since formalin fixation completely inactivates endogenous tissue RNAses.
While the length of fixation and the temperature of the paraffin during embedding can also negatively affect the ability to extract and the quality of the RNA, the storage itself of FFPE tissues under appropriate temperature and humidity conditions has very little additional negative effect on RNA integrity if it is limited to 1 year. Beyond 1 year of storage, further RNA damage is to be expected. 19
The main question then becomes the following: how can the damage to nucleic acids during the FFPE process, if unavoidable, at least be reduced? Maximizing the detection potential of nucleic acids in FFPE tissues starts with the implementation of good laboratory practices for tissue collection, handling, and processing. Once an appropriate tissue has been selected, it is important to minimize the interval between collection and fixation. In surgical pathology, this interval is commonly referred to as cold ischemia time. Specimens are typically kept either at room temperature or in a refrigerator during this time. It is clear that both nucleic acid and protein changes take place during the warm ischemia time, which is the time a tissue remains at body temperature after its blood supply has been reduced or cut off. Its duration should be minimized and, whenever possible, also recorded.
Based on the fact that the rate of penetration of formalin into tissues is approximately 1 mm/h, it is important that tissues be properly sectioned to the appropriate thickness prior to fixation. A thickness of 0.5 to 1 cm is desirable.
The quality of the formalin used for fixation is crucial. As mentioned above, formalin that has not been buffered to neutral pH can result in nucleic acid fragmentation. Unbuffered formalin solutions also degrade fairly rapidly, whereas neutral buffered formalin (NBF) has a fairly long shelf life. Several formalin buffering systems have been described. Phosphate is superior to other buffering systems in terms of both quality and yield of the nucleic acids obtainable from FFPE tissues. 19,20
The volume/weight ratio of NBF to tissue and fixation times are other elements that have to be monitored. The proper fixative to tissue ratio is approximately 10:1. Proper fixation times range from 12 to 36 hours. 19 Tissue fixation times beyond 36 hours have been reported to have a negative impact on the quality of the nucleic acids to be analyzed. 19 It is therefore essential to be familiar with the workflow of the histology laboratory embedding the tissue. Since routine tissue processing commonly takes between 11 and 13 hours, many routine laboratories will run their tissue processors overnight. Laboratories that have no weekend service will commonly retain tissues in the processor within formalin, thereby effectively prolonging formalin fixation by 48 hours. In such cases, or whenever the tissue processing is expected to be delayed, superior sample quality can still be ensured by transferring the completely formalin-fixed tissue after 24 hours into 60% ethanol prior to tissue processing. 19 The influence of the length of fixation on RNA quality has been reviewed previously. 1
Elements of the paraffin embedding process can also affect nucleic acid availability. Both the quality of the paraffin used and the embedding temperature can play a role. It is generally recommended that synthetic rather than natural paraffins be used. 19,20 Since elevated temperatures could also lead to nucleic acid damage, the embedding temperature should be kept below 60°C.
Once embedded, blocks should be stored under controlled temperature and humidity conditions. As indicated above, the additive damage resulting purely from storage is only obvious after the first year of storage, especially when storage occurs under suboptimal conditions. In terms of RNA preservation, storage of tissues as tissue blocks is superior to storage in the form of tissue sections. 110 Covering the tissue surface with a layer of paraffin will further protect nucleic acids.
Overall, FFPE tissues cannot be expected to yield the quality or quantity of nucleic acids obtainable from unfixed tissues. However, proper tissue collection, handling, and processing procedures will positively influence both the quality and quantity of nucleic acids that are available for downstream extraction procedures.
Nucleic Acid Extraction
Nucleic acid extraction from FFPE consists of 3 steps: deparaffinization, digestion, and purification. For deparaffinization, treatment with xylene or a xylene replacement such as Citro-Solv is most commonly used. Steinau et al 119 demonstrated that high-heat treatment had an advantage over xylene-based deparaffinization in extracting human papillomavirus 16 (HPV16) DNA. The main positive effect noticed was on viral DNA yield. By adding heat treatment to the commercially available kit protocols, HPV DNA could be extracted efficiently from FFPE tissues. The recommended method allowed use of a limited amount of starting material, was less laborious, and reduced the risk of tissue loss during the washing steps. Validation included its use on a total of 1500 blocks contributed by a number of different pathology laboratories and prior successful use at the authors’ laboratory on about 5000 blocks.
Proteinase K treatment is an essential component of the DNA extraction process because it is responsible for liberating the protein-bound DNA and making it available for subsequent amplification. Elements that influence the effectiveness of the proteinase K digestion are the thickness of the tissue slices being digested, the concentration of proteinase K, and the length of digestion.
RNA degradation and modification in FFPE tissues is as much of an issue as it is for DNA, and the amount of mRNA that can be extracted from FFPE tissues will invariably be lower than the amount extracted from fresh tissues. von Ahlfen et al 134 reviewed various determinants affecting the quality of RNA obtainable from FFPE tissues. As is the case for DNA, proteinase K digestion is a crucial part of the extraction protocol to maximize mRNA yield. Proteinase K treatment results in the digestion of the proteins that are cross-linked to RNA to the level of tetrapeptides 62 but has no effect on the methylene bridge that actually forms the cross-link. In contrast, the subsequent heating step breaks the actual cross-links but has to be limited to avoid heat-induced RNA damage. 77 Chaotropic substances used as substitutes to proteinase K treatment have been shown to have far lower efficiency. 77
Chung and Hewitt 20 emphasized that the RNA extraction protocol is crucial for the quality and quantity of RNA obtained from FFPE tissues. In the protocol they described, tissues were deparaffinized at 95°C for 15 minutes and subsequently lysed for 3 days at 65°C. Even though the yield and purity of the RNA obtained with these modifications were high, the fragment length was still limited to 200 to 300 nucleotides.
The challenge of recovering viral RNA from FFPE tissues that had been fixed for 3 to 4 weeks was discussed by McKinney et al. 79 Two different commercial kits were used for RNA extraction. The overall protocol modifications introduced by these authors consisted of increasing the deparaffinization temperature to 65°C, increasing the proteinase K concentration to 2 mg/ml, and increasing the proteinase K treatment time to 24 hours. The results indicated that it was not possible to extract RNA from FFPE tissues using commercial kits without modification. Among the modifications made, increasing the proteinase K digestion time from 2 to 24 hours was essential to obtain sufficient amplifiable RNA.
Bonin et al 9 compared the efficiency of various DNA and RNA extraction kits. For DNA extraction, both commercial kits and in-house methods were satisfactory. The choice between methods that included DNA purification versus those that only extracted DNA was based on the downstream assay that had to be performed on the extracted DNA. Among the different RNA extraction protocols tested, commercial silica-based extraction kits were superior to commercial or in-house phenol extraction-isopropanol precipitation methods, both in terms of RNA quantity and quality.
PCR-Based Assays to Amplify DNA or RNA Extracted From FFPE Tissues
PCR-based assays are commonly used to amplify nucleic acids extracted from FFPE tissues. RNA detection by PCR, which first requires conversion of the RNA target to complementary DNA (cDNA) by reverse transcription, is referred to as RT-PCR. The 2 main detection formats of amplicons are gel-based or real-time PCR. In gel-based assays, amplicons are size-separated by agarose-gel electrophoresis. They are labeled, either during or after electrophoresis, with a fluorescent dye and then visualized under UV light. In real-time PCR assays, evidence of amplicon generation is detected and recorded during the assay itself (ie, in “real time”). One of the 2 common formats is based on the incorporation of a fluorescent dye, such as SYBR Green or equivalent, as evidence of amplicon accumulation. In the other format, referred to as “Taqman” or other probe-based assay, a synthetic oligonucleotide probe is incorporated into the reaction mixture. The principle of this assay format is depicted in Figure 1. The probe is labeled at its 5′ end with a fluorophore and at its 3′ end with a fluorescence quencher, which prevents the generation of a fluorescent signal as long as the probe is intact. Assuming that a specific target is present in the extracted sample, DNA polymerase-induced probe degradation takes place during each cycle as part of template strand copying. This probe degradation “liberates” the fluorophore, which then generates the fluorescent signal being quantitated. A specialized thermocycler incorporating a fluorometer is essential for conducting real-time PCR assays in either format. The basics of PCR and its varied formats have been reviewed extensively. 100,107,115,151
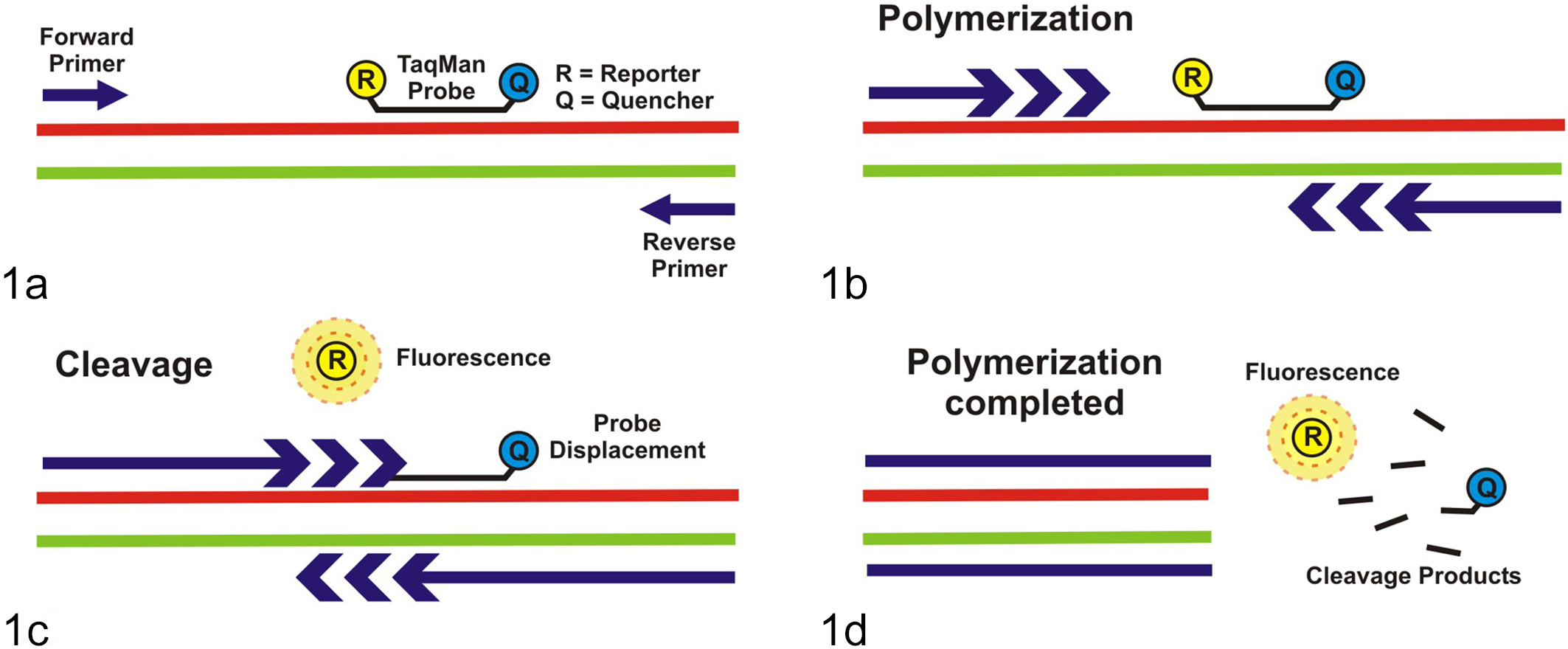
Taqman probe-based real-time polymerase chain reaction (PCR). (a) Primers and probe anneal to the complementary target sequence; the probe, which is an oligonucleotide containing a fluorophore (R) and a fluorescence quencher (Q), is still intact. (b) Polymerization takes place during the extension step of each PCR cycle. (c) The fluorophore (R) is released from the Taqman probe, mediated by the 5′-exonuclease activity associated with the DNA polymerase as it copies the template strand and displaces the probe. The fluorophore (R) is no longer quenched by the quencher (Q) and fluoresces. (d) Fluorescence accumulates in the reaction mix as polymerization is completed at every PCR cycle and is detected in real time.
DNA extracted from FFPE tissues will typically have a size limit of 300 to 400 base pairs (bp), and attempts to amplify targets over 600 bp should be avoided. Various studies have shown that RNA extracted from FFPE samples is often less than 200 nucleotides long. 23,97,134 Greer et al 37 compared the effects of fixatives and the length of storage on the efficiency of downstream target detection by PCR. They demonstrated that after 8 days of fixation in NBF, targets that were 536 bp in size were amplified, but those that were 989 bp in length were barely detectable. After 1 month of fixation, the maximum detectable target size dropped to 268 bp. Fixation with other fixatives, such as omnifix, acetone, and ethanol, allowed detection of larger DNA targets, either at 8 or 30 days postfixation. Therefore, it is important to select an assay format capable of amplifying targets that are limited in size but retain high sensitivity and specificity. Probe-based real-time PCR readily accommodates this requirement, especially for the more compromised RNA templates. Targets even as small as 87 nucleotides do not create problems because postamplification visualization of the actual product is no longer needed.
The potential for laboratory contamination, which can lead to false-positive results, is another issue to take into account in assay format selection. Even in a well-designed laboratory, the chance for laboratory contamination increases when the number of assays run on a given day is high. Laboratory contamination is less of a concern with real-time PCR assays, since the laboratory environment is not being exposed to high copy numbers of PCR amplicons, especially during gel loading.
Inhibition of amplification by substances present in the nucleic acid extract is always a potential issue associated with PCR assays. Inhibitory substances can be part of the sample itself (eg, hemoglobin). PCR inhibition can also be the result of the extraction process (eg, residual xylene or ethanol used for nucleic acid precipitation). The use of input nucleic acid amounts that are too high also has a negative effect on PCR that is referred to as template inhibition. Since the end result of the uncontrolled presence of inhibitors is the potential for false-negative results, careful monitoring for inhibition needs to be incorporated into PCR assays. A commonly used approach for monitoring is the coamplification of an internal control. This can, for example, be a synthetic construct with primer binding sites that are identical to those of the authentic target. Successful amplification of the internal control generates an amplicon that can be size-differentiated from the authentic target in gel-based assays. Alternatively, the internal control can be detected in a real-time assay by using a probe labeled with a different fluorophore.
Votavova et al 135 described the use of real-time PCR as the analytical method to detect infectious agents in FFPE tissues and comprehensively examined the influence of RNA extraction, reverse transcription, and the fluorescence detection system on the performance of this detection method. A commercial kit was the most efficient extraction method. SYBR Green and Taqman real-time PCR assays were compared in terms of amplification efficiency. Primer-primer annealing interfered with the interpretation of SYBR Green assay results.
Alternatives to Formalin Fixation
As stated earlier, fixation of tissues in formalin results in excellent preservation of morphological detail; however, formalin has a rather detrimental effect on nucleic acids, thus lowering their detection by various molecular methods. Acceptable alternatives to formalin fixation should retain morphological detail needed for histological examination but at the same time improve upon nucleic acid preservation.
The HOPE fixative has been proposed as an alternative to formalin. By real-time PCR, the detection limit of Mycobacterium tuberculosis DNA was 110 times lower than with formalin fixative. 133 Another promising method makes use of a combination of the so-called molecular fixative (MF), a microbicidal mixture of methanol and polyethylene glycol, and a rapid tissue processor with microwave technology. 22,89,124,131 This system is reported to enable optimal preservation of tissue morphology and nuclear detail as well as preservation of the integrity of DNA, RNA, and proteins in paraffin blocks. 22,89,131 The efficiency of DNA and RNA detection by in situ hybridization and real-time PCR from cells or tissues fixed with 9 different fixatives was described by Yan et al. 150 From the overview table presented in their article, it is clear that both the substrate and the downstream assay determine the type of fixative that is most suitable. For example, overnight fixation in 10% NBF was either excellent (on cells) or acceptable (on tissue) when the downstream assay was in situ hybridization. On the other hand, this fixation method was scored as unacceptable for PCR-based detection of DNA or microRNA. Acetone and methanol were clearly the best fixatives when solution phase PCR assays needed to be performed on fixed tissue. An excellent overview of fixatives that are potential alternatives to formalin is also presented in the recent review by Klopfleisch et al. 60
In summary, alternatives to formalin are available and in fact may be preferred in preplanned projects involving liquid phase molecular detection assays. In contrast, in many situations in which molecular nucleic acid detection assays are added to routine histopathological examination, nucleic acid extraction will have to be done from FFPE tissues.
Application of Molecular Tools for Viral Disease Discovery and Diagnosis
Discovering Viruses Using Consensus PCR
Consensus PCR is a very useful tool for detecting novel agents belonging to a particular virus family. This is achieved by designing broadly reactive primers, based on known highly conserved regions of a gene within the virus family that is targeted. Most of these PCR assays are of the gel-based type. Some are of the nested format to maximize the sensitivity of the test. Nested refers to the fact that the assay consists of 2 rounds of PCR. The second round PCR is done with a primer set that is internal to the set used in the first round. The suspect amplicon that meets size expectation is then excised from the gel, purified, and sequenced for target authenticity verification, typically by performing Basic Local Alignment Search Tool (BLAST) searches of the databases with the sequences obtained. The following are examples of these consensus primer-based PCR assays that we have found useful for disease investigations, particularly for the identification of novel and emerging viral agents.
Herpesviruses
When investigating the possibility of a herpesvirus infection, the consensus herpesvirus PCR by VanDevanter et al 128 works well with FFPE tissue DNA extracts. This assay makes use of a nested primer set that targets a conserved region of the DNA polymerase gene shared by all 3 herpesvirus subfamilies (alpha-, beta-, and gammaherpesvirus). This assay was initially validated by its ability to detect 21 herpesvirus species (8 human and 13 animal viruses). 128
Other investigators have successfully used this consensus PCR assay to identify novel herpesviruses. Kleiboeker et al 59 used FFPE lung tissues to investigate the presence of herpesvirus infections in donkeys with interstitial pneumonia. Williams et al 144 investigated whether a herpesvirus is associated with equine multinodular pulmonary fibrosis (EMPF), a newly described disease in horses. The consensus herpesvirus PCR was initially used to screen lung samples (fresh frozen or FFPE) from 24 horses. Sequencing of the PCR amplicon from the index case showed the presence of a herpesvirus DNA polymerase sequence with 98% similarity to equine herpesvirus (EHV) 5. Follow-up testing with an EHV5-specific PCR, amplifying a 344-bp target, demonstrated that all 24 horses were positive. Only 8 of the 13 FFPE lung samples were positive by the consensus PCR, while all 13 tested positive with the EHV5-specific assay. It is important to bear in mind that in most cases, specific PCR assays are more sensitive than their generic counterpart. Nevertheless, the VanDevanter consensus assay was reported to have a sensitivity that can detect levels at or below 100 genome equivalents per 100 ng of DNA. 128
The consensus PCR was also used in our laboratory to detect a herpesvirus from FFPE brain tissue from a polar bear that exhibited neurologic signs. 28,113 The sequence of the consensus PCR product showed a 98% identity to EHV1. The polar bear specimen was also positive by a real-time EHV1-specific PCR. Upon further sequencing of the DNA polymerase and the glycoprotein B gene, the herpesvirus was determined to be EHV9. The EHV1-specific PCR was positive in this case due to the high sequence similarity between EHV9 and EHV1.
Papillomaviruses
Consensus PCR assays for papillomaviruses are highly useful tools for detecting novel viruses associated with benign or malignant neoplastic diseases of the skin and mucosal membranes both in humans and animals. In humans, more than 100 papillomaviruses have been identified, molecularly characterized, and grouped into 5 genera (alpha-, beta-, gamma-, mu-, and nupapillomavirus). 25,64 Preneoplastic and neoplastic skin diseases associated with papillomavirus infection have been documented in a variety of animal species. 91 Targeted genes for the generic detection of papillomaviruses are L1 and E1, due to the presence of highly conserved regions within both of these genes.
The primer pair MY09/MY11 targets a 450-bp region of the L1 gene. 74 Animal papillomaviruses that have been detected with the MY09/MY11 primers include bovine papillomaviruses 96 and, very recently, a baboon papillomavirus. 8 The L1 gene-targeted assay 74 is also applicable to FFPE tissue samples. In 2006, we reported the detection and identification of a novel papillomavirus in an Egyptian fruit bat (Fig. 2). 80 This bat papillomavirus has subsequently been completely sequenced and genetically characterized. 108
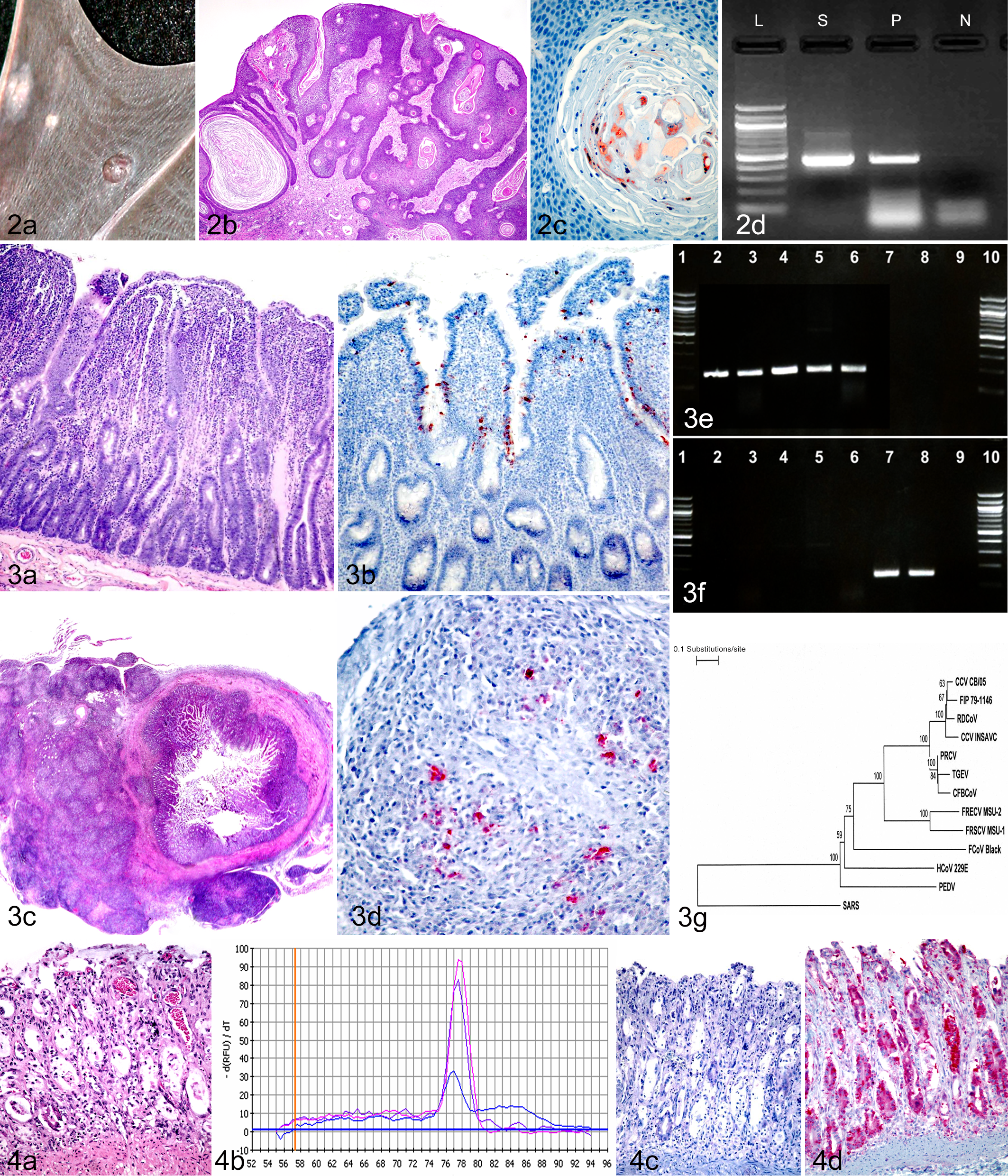
Basosquamous carcinomas caused by a chiropteran papillomavirus in an Egyptian fruit bat (Rousettus aegyptiacus) (RaPV-1). (a) Wing membrane with multiple raised, slightly irregular neoplastic masses. (b) Thick anastomosing rete pegs of basilar neoplastic epithelial cells extend from the overlying hyperplastic epithelium into the underlying dermis. Hematoxylin and eosin (HE). (c) Positive immunohistochemical labeling for papillomavirus antigen (brown) in nuclei of dyskeratotic cells within central zones of keratinization. AEC chromogen, hematoxylin counterstain. (d) Polymerase chain reaction (PCR) using degenerate primers designed to amplify a 450–base pair (bp) segment of the L1 gene of the human papillomavirus genome detected a target within neoplastic tissue. L, ladder; N, negative control; P, positive control; S, sample of neoplastic tissue.
The E1 gene is another good target for consensus papillomavirus PCR. Iftner et al 46 used this broad-range PCR, capable of amplifying at least 64 different human papillomavirus types, to detect papillomavirus in nonmelanoma skin cancers of nonimmunosuppressed individuals. Nespeca et al 94 applied this assay, in a multiplex format, to detect papillomavirus DNA in FFPE squamous cell carcinoma (SCC) tumor specimens from cats.
Following the same multiplex format of the E1 assay, we were able to amplify papillomavirus sequence from wart-like paw pad lesions in dogs 5 and from oral and genital masses collected from wild spotted hyenas in Kenya. 92 In the canine study, FFPE biopsy specimens from 11 paw pad lesions, 6 of which were obtained from Greyhounds and 5 from dogs of other breeds, were evaluated by PCR, immunohistochemistry (IHC), and histologically for the presence of viral inclusion bodies. All Greyhound samples were negative for papillomavirus, while 3 of the 5 non-Greyhound lesions were virus positive by both IHC and the consensus PCR. Two of these also showed viral inclusion bodies. The results suggested that wart-like paw pad lesions in dogs other than Greyhounds may potentially be associated with papillomavirus infection.
In the hyena study, microscopic lesions in the biopsy samples were consistent with papilloma virus infection. Papillomavirus sequence was amplified from the papillomatous lesions with the E1 gene PCR. Follow-up full-genome sequencing confirmed it to be the first papillomavirus identified in a species belonging to the Hyaenidae family. 120
Coronaviruses
Within the past decade, coronavirus has been implicated as the causative or associated agent in several newly emerging infectious diseases in humans and animals. Novel human coronaviruses (HCoV) include the severe acute respiratory syndrome (SARS) virus, HCoV-NL63, and CoV-HKU1. 29,34,63,101,126,147 Emerging coronaviral diseases in animals include epizootic catarrhal enteritis 143,146 and feline infectious peritonitis (FIP)–like systemic disease in ferrets, 36,54,75,145 a fatal systemic disease in dogs, 12 and mink epizootic catarrhal gastroenteritis. 132
When a coronavirus etiology is suspected, pan-coronavirus detection methods are very useful to the diagnostician. One such tool is the consensus coronavirus RT-PCR assay, developed by Escutenaire et al. 32 This RT-PCR assay targets a 179-bp region of the open reading frame (ORF) 1b of the replicase gene. The assay, designed for real-time detection using SYBR Green chemistry, was validated with a mix of 36 human or animal coronavirus strains, covering the alpha-, beta-, and gammacoronavirus genera. We previously used this RT-PCR, in the gel-based format, to detect coronavirus RNA in frozen or archived FFPE tissues collected from ferrets diagnosed with the FIP-like systemic disease (A. G. Wise, R. K. Maes, and M. Kiupel, unpublished data). 36 This systemic ferret coronavirus was recently genetically characterized and compared with the enteric ferret coronavirus (Fig. 3). 145 The positive PCR results with FFPE samples imply that the target template, being only 179 nucleotides in size, had remained intact in sufficient quantity for detection, despite sample fixation and embedding.
Based on our experience, both extraction yield and quality of RNA from FFPE tissues were higher when commercial RNA extraction kits, developed specifically for FFPE samples, were used. The protocol for these specialized kits consistently includes an incubation step at 80°C, intended to partially reverse formalin-induced protein cross-linking to the nucleic acids. In this heating step, it is important to strictly observe the recommended incubation time to avoid heat-induced RNA damage. 134 As a general rule, however, when testing for RNA viruses by PCR, it is still best to use fresh/frozen samples when available.
Polyomaviruses
Polyomaviruses are known to cause subclinical infections with lifelong persistence in immunocompetent mammalian hosts. This is in contrast to avian polyomaviruses (eg, budgerigar polyomavirus and goose hemorrhagic polyomavirus), which cause acute and fatal disease in apparently healthy parrot fledglings and goslings. 52 In the immunocompromised mammalian host, opportunistic reactivation of the virus can result in severe clinical disease. In humans, JC virus and BK virus are the causative agents of progressive multifocal leukoencephalopathy and polyomavirus-associated nephropathy, respectively. 21,99 A number of novel animal polyomaviruses have recently been identified through the use of a nested broad-spectrum PCR assay developed by Johne et al. 51 This consensus assay targets the conserved regions of the polyomavirus VP1 gene. With this assay, novel polyomaviruses were detected from fresh tissues, blood, or feces collected from a chimpanzee, 51 a squirrel monkey, 130 bats of Canada, 85 Bornean and Sumatran orangutans, 39 and wild rodents from Zambia. 98
The consensus PCR also worked well for the detection of polyomavirus in FFPE samples, despite the product size target (>600 bp) of the first-round PCR. A canary polyomavirus was detected in FFPE tissues from fatally diseased canaries. 43 A novel polyomavirus was detected in FFPE placental tissue of a northern fur seal in Alaska. 30 In a recent report, this VP1 gene-targeted PCR was used to detect a polyomavirus in FFPE kidney samples from Standardbred horses with nephritis reminiscent of BK polyomavirus nephropathy in humans. 49 The VP1 protein sequence of this virus was identical to that of the equine polyomavirus that was isolated from the eyes of horses by another group of investigators. 109
Differentiating Virus Strains by Detection of Molecular Signatures
PCR is a versatile tool that can be designed to differentiate strains according to genotype or nucleotide sequence differences that are consistent between strains of varying pathotypes or tissue tropisms. PCR can also be used to discriminate a field strain from a vaccine strain. This is made possible through the design of primers and/or probes tailored to recognize specific genetic signatures of strains. Our laboratory recently provided preliminary evidence that ferret systemic coronavirus (FRSCV) can be differentiated from ferret enteric coronavirus (FRECV) based on a significant sequence difference in the spike (S) gene. 145 The molecular signatures observed were consistent in 3 strains analyzed for each pathotype. In the same study, we developed 2 S-gene genotype-specific RT-PCR assays by taking advantage of the sequence difference observed between the 2 pathotypes of ferret coronavirus (Fig. 3E,F). In this case, primer design is crucial to obtain the specificity of the assay. These are traditional gel-based assays with primers that bind to very short targets: 157 bp for FRSCV and 147 bp for FRECV. These short PCR targets fall within the ideal size range when working with RNA extracted from FFPE samples.
Even a single-nucleotide change that is found consistently in 2 types of strains (eg, wild type vs vaccine) can be taken advantage of in the design of diagnostic molecular assays. In this scenario, the discrimination is made possible via Taqman-based real-time PCR where probe design offers the specificity and the primers bind to a conserved target shared by all strains of the virus. An example of this PCR approach is the assay designed by Decaro et al 26 to differentiate between canine parvovirus (CPV) type 2b vaccinal and field strains. In designing the probes, the authors took into account the consistent “A-to-T” base substitution in the capsid gene that allows the discrimination between vaccine and field strain. Hence, 2 probes that differ in sequence by only 1 nucleotide are each labeled with a differentiating fluorophore (eg, FAM or VIC) and simultaneously used in 1 PCR reaction. The amplicon size for this assay is only 85 bp, making it quite suitable for testing archived FFPE tongue specimens, which are excellent complementary samples for parvoviral testing in dogs and cats. 81
Yet another way to differentiate vaccine virus from a wild-type strain is by using a combination of PCR and follow-up sequencing analysis. One example is the differentiation of virulent canine distemper virus (CDV) from the 2 seed strains commonly used in the preparation of commercial vaccines. It has been shown that CDV field strains exhibit up to 13 conserved nucleotide changes in a region of the P gene in comparison to the 2 vaccine strains that are identical. 44,72 The primers for these assays target either a 429-bp 6 or 585-bp 72 fragment of the P gene. It is much more difficult to conduct these assays successfully on RNA extracted from FFPE specimens due to the relatively large amplicon size. Nevertheless, in our experience, if the tissue sample has a very high viral load (with real-time RT-PCR cycle thresholds <20) to begin with, it is still possible to detect >500-bp fragments of RNA in FFPE tissues.
The examples discussed in this review specifically pertain to the discovery of viral agents. However, it is clear from existing literature that the approaches described here are also applicable to the identification or discovery of a broad range of other infectious agents. The main principles that need to be adhered to, regardless of the target, are proper specimen selection and preservation, fixative type and fixation time, optimum extraction method, and a molecular assay that is optimized to detect a relatively small target, since target degradation and modification are to be expected in FFPE tissues.
Differentiating Infection vs Disease
While PCR is a sensitive method to detect viral nucleic acid, care has to be taken not to automatically interpret detection of an infectious agent as the cause of a disease process. The use of PCR for FIP diagnostics is an example. PCR only detects coronavirus RNA and does not differentiate between enteric and systemic feline coronavirus. Thus, this test should not be used in isolation but should be supplemented by clinical, gross, and histologic findings, and immunohistochemical demonstration of coronavirus antigen within intralesional macrophages should be used to confirm a diagnosis of FIP.
In other cases, detection by PCR of a target within tissue, even within a tissue with characteristic pathology, can be insufficient to differentiate between infection and replication of a virus. Consider the following example. A 15-month-old female Shih Tzu with a history of mucopurulent ocular discharge, vomiting, hemorrhagic diarrhea, and neurologic deficits was euthanized and submitted for a complete necropsy. The dog had recently been vaccinated against CDV and CPV-2. At necropsy, the primary lesion was observed in the gastrointestinal tract. Throughout the length of the small and large intestines, the luminal contents were semisolid to loose and watery and variably hemorrhagic. The duodenal and jejunal mucosa had numerous petechiae. Histologically, lesions were limited to the intestines. Within multiple intestinal segments, there was enteritis centered on mucosal crypts. Affected crypts displayed a variety of changes, including mild to marked ectasia, segmental to circumferential epithelial attenuation, loss, and regeneration (Fig. 4A). Crypt lumina variably contained necrotic cellular debris (crypt abscesses) and/or amphophilic flocculent material (mucin). The villous lamina propria was infrequently expanded by hemorrhage. No infectious organisms or intracellular inclusions were observed. A diagnosis of subacute, segmental, hemorrhagic enteritis with crypt epithelial attenuation, necrosis, and regeneration was given, and parvovirus infection was suspected. PCR for CPV-2 was performed on sections of small intestine and gave a strong positive result (Fig. 4B). Based on gross, microscopic, and PCR results, a diagnosis of canine parvovirus infection was originally made, especially since intestinal crypt necrosis is one of the hallmarks of canine parvoviral disease and the absence of pulmonary or nervous tissue lesions made an infection with CDV less likely. However, in addition, immunohistochemical testing for canine parvovirus was performed on sections of the small intestine. The IHC was negative in 2 repeats (Fig. 4C). At this point, additional RT-PCR and IHC for CDV were performed on sections of the small intestine. Viral nucleic acid was detected by RT-PCR, and positive immunolabeling for CDV antigen was identified in areas of enteric inflammation and, specifically, within epithelial cells of necrotic crypts (Fig. 4D). Following sequence analysis of PCR amplicons, CDV was confirmed as a field strain and not a vaccine strain, thereby confirming a postvaccination CDV infection as the cause of the described lesions. Although sequencing of the parvovirus was not performed, the positive PCR result for CPV-2 in this case was likely due to limited replication of virus after recent vaccination, rather than infection with a CPV-2 field strain. This case illustrates that basing a diagnosis on histopathology and PCR results alone, without knowledge of the vaccine status or sequence analysis of the PCR amplicons, can easily lead to the wrong conclusion. Demonstration of viral antigen by fluorescent antibody or IHC within microscopic lesions is the preferable diagnostic approach in such cases. 81
Molecular Tools for Diagnosing and Predicting Prognosis of Neoplastic Diseases
PCR to Detect Antigen Receptor Rearrangements
Detection of clonal expansion, representing the hallmark of cancer, has been proven to be clinically useful for the diagnosis of lymphoma based on the detection of specific and dominant T-cell receptor and/or immunoglobulin gene arrangements in both animals and humans. When histology and immunohistochemistry cannot distinguish between a reactive (polyclonal) population and a neoplastic (clonal) population of lymphocytes, PCR to detect antigen receptor rearrangements (PARR) can be helpful for both B- and T-cell lymphomas (Fig. 5). PARR testing to determine clonality of lymphocytic proliferations has recently become a common practice in canine and feline medicine. This assay has been used clinically in many different situations, such as distinguishing between canine indolent nodular lymphoma and atypical lymphoid hyperplasia; 125 intestinal lymphoma and inflammatory bowel disease (IBD); 7,58,87,88,122 hepatic lymphoma and lymphocytic cholangitis/cholangiohepatitis; 121,136 leukemia and reactive, nonneoplastic lymphoproliferative conditions; 129,149 and cutaneous lymphoma and lymphocytosis, erythema multiforme, or other lymphocytic inflammation. 2,86,93
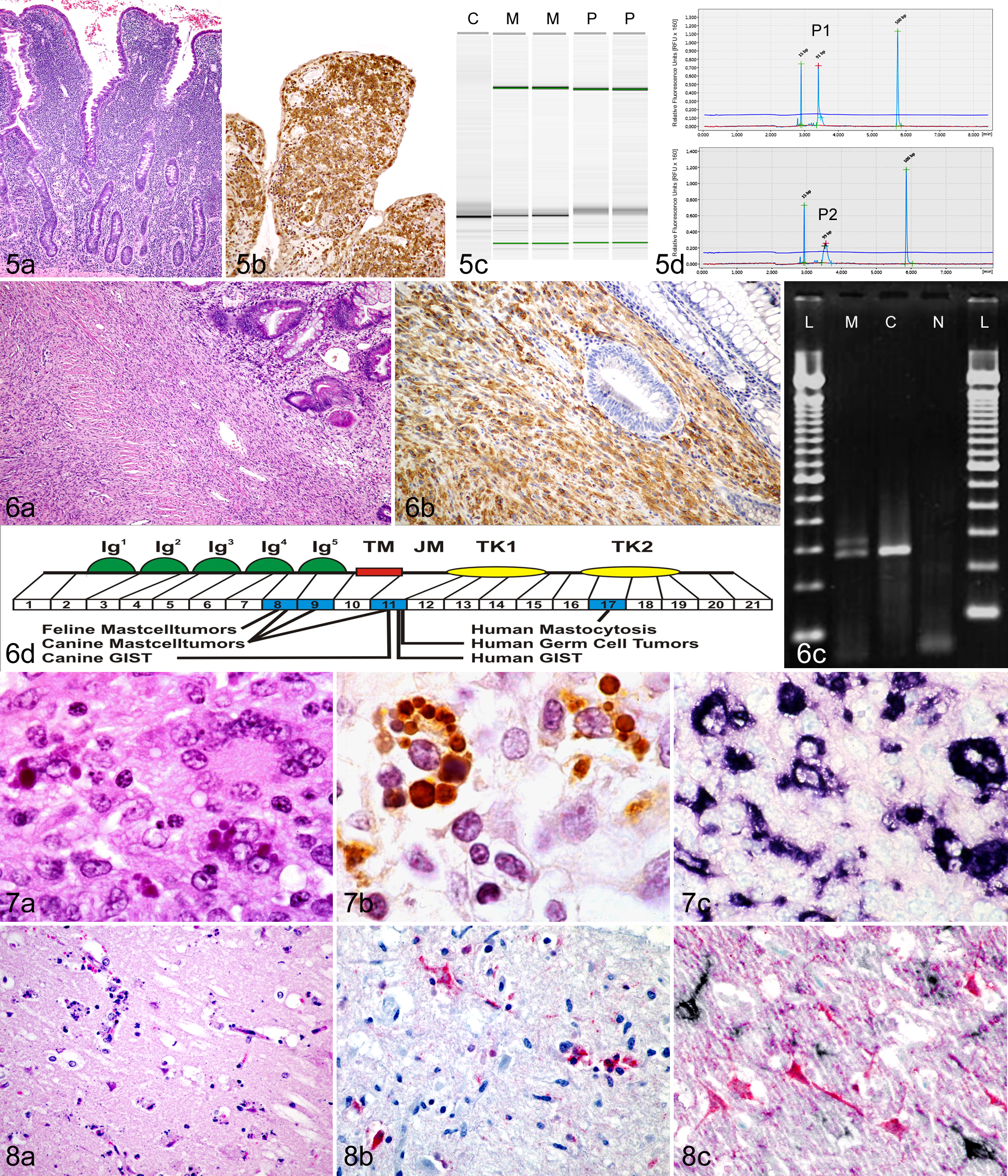
Differentiating enteropathy-associated T-cell lymphoma (EATL) from inflammatory bowel disease in cats. (a) EATL with diffuse infiltration of the mucosal lamina propria by small neoplastic T lymphocytes. Hematoxylin and eosin (HE). (b) Neoplastic cells that are diffusely immunohistochemically positive for CD3 (brown) expand the lamina propria and form intraepithelial plaques. DAB chromogen, hematoxylin counterstain. (c) Polymerase chain reaction (PCR) for antigen receptor rearrangement for T-cell receptor γ. Capillary gel electrophoresis results in single strong bands for monoclonal (neoplastic) cell populations (lanes M), while polyclonal (inflammatory) cell populations are characterized by a smear (lanes P). Lane C, monoclonal positive control. (d) Similarly, monoclonal (neoplastic) cell populations are characterized by a single dominant narrow peak (P1) in the electropherogram, while polyclonal (inflammatory) cell populations produce more of a curve or broader peak that is at least 3 times shorter (P2). Size markers are represented as peaks at 15 base pairs (bp) and 500 bp in the electropherogram and green lines in the PCR gel.
Neoplastic lymphocytes of a particular neoplasm contain unique DNA regions that are found primarily within the CDR3 region of both the immunoglobulin heavy chain gene for B cells and the T-cell receptor (TCR) γ chain gene for T cells. These regions encode the antigen binding region of each receptor, which is formed by the recombination of V, D, and J segments in B cells and by the recombination of V and J segments in T cells. Primers that are specific for conserved regions of V and J segments can be used to amplify CDR3. Then, the PCR products are separated by size and, if there is a proliferating lymphocyte population with a single-sized receptor (clonal population), a dominant band will be present in the gel view (Fig. 5C) or a dominant peak will be present in the histogram view (Fig. 5D). 13
One of the PARR assay’s most common applications in diagnostic practice is to differentiate between IBD and intestinal lymphoma in cats. Specific histologic features that aid in this distinction have been well established in cats. 10,58,87 However, in many cases, histology alone or histology plus immunophenotyping is insufficient for diagnosis. PARR testing is especially helpful when only superficial endoscopic biopsy samples are available or in early cases of lymphoma that lack submucosal invasion or marked epitheliotropism (Fig. 5). A diagnostic algorithm, using histologic features, immunophenotyping, and PARR testing, has been established to aid in the diagnosis of feline intestinal lymphoma. 58
Just like many of the molecular tools described in this review, PARR testing should never be used as a stand-alone test. The pathologist needs to interpret the histologic features, immunophenotyping, and clinical findings in conjunction with one another. There are several reasons for this. First, the sensitivity of the primers to detect rearranged immunoglobulin heavy chain (IgH) or TCR genes must be taken into consideration. Qualitative sensitivity is defined as the ability of the PARR assay to detect a purely clonal population of lymphocytes. 118 Quantitative or analytical sensitivity is defined as the assay’s ability to detect a clonal rearrangement in a background of nonneoplastic lymphocytes. 55 In some cases, a neoplastic population of lymphocytes is present within an inflamed tissue, and the clonal lymphocytes can be, in effect, “diluted out” by the polyclonal inflammatory cells. Thus, different tissues may also influence the sensitivity of the assay if they are more prone to having a concurrent inflammatory or reactive population of lymphocytes. For example, PARR testing has a higher sensitivity in nonlymphoid tissues compared with lymphoid tissues. 13 It has been speculated that “the rearranged sequences from normal lymphocytes compete with amplification of the neoplastic DNA.” 13 We routinely receive blocks of tissue or unstained slides for clonality testing that contain multiple organs, such as intestine, liver, and lymph node. To increase the quantitative sensitivity of the PARR test in these cases, we only use paraffin shavings that contain the tissue of concern.
The first clonality assays that were developed to detect clonal populations of T lymphocytes in dogs were based on limited sequence data, and therefore they often miss certain V and J segment rearrangments. 55 Based on more recent studies that have identified additional less commonly expressed rearrangements and provided more detailed characterization of the complete canine TRC locus, 56,76,149 a new multiplex PARR assay has been developed for assessment of canine T-cell proliferations that has been shown to have an improved sensitivity. 55 In addition to using multiplex primers, the sensitivity of the PARR assay has also been improved by using capillary gel electrophoresis. 66 Multiplex primers generate more complex electrophoretic gels and, therefore, capillary gel electrophoresis is particularly important to accurately interpret the more complex gel patterns; conventional gels may not provide sufficient resolution to identify clonal bands in a polyclonal background. 66 Furthermore, knowledge of primer design and expected bands is crucial to avoid interpretation errors.
False-negative results can occur in some cases of lymphoma that are immunophenotypically of either B- or T-cell origin but do not have a rearrangement in their respective gene, IgH or TCR. For example, some B-cell lymphomas may have a polyclonal result with PARR testing for rearrangements in IgH but may actually have a rearrangement in TCR. This is an example of lineage infidelity. If only PARR testing for IgH rearrangement is performed, cases such as these may be misdiagnosed as reactive or inflammatory rather than neoplastic. Thus, for some cases that are suspected to be neoplastic, PARR testing for both B- and T-cell clonality is required. Also, in some cases of canine and feline lymphoma, dual clonality has been reported. 58,125 This can occur when a lymphoma has a dual genotype with lineage infidelity or, rarely, when a composite lymphoma is present, which is defined as concurrence of 2 monoclonal malignant B- and T-cell populations. 35 Laser capture microdissection can be used to separate specific cell populations for clonality testing, but this is a costly method that is not routinely available at most veterinary laboratories. It is important to remember that the immunophenotyping defines the type of lymphoma as B or T cell, not the gene rearrangement. Thus, in the example above, the diagnosis would be a B-cell lymphoma with TCR gene rearrangement.
Specificity of the PARR assay also needs to be considered. Specificity is defined as a test’s ability to correctly identify negative results. In general, most PARR assays for assessment of feline and canine B-cell and T-cell proliferations are highly specific. Sample size is one factor that may influence specificity. For example, a very small endoscopic intestinal biopsy with a poorly cellular inflammatory lymphocytic infiltrate may result in a clonal PCR result. It is possible that such samples with very small numbers of lymphocytes may contain nonneoplastic, reactive lymphocytes that were derived from the same precursor cell. Another instance in which a false-positive result can occasionally occur in dogs is in cases of Ehrlichia canis infection. E. canis infection has been reported to produce a clonal expansion of T lymphocytes. 13,129 It is also important to remember that not all clonal populations are malignant. For example, a canine cutaneous plasmacytoma can have a clonal immunoglobulin rearrangement, but these tumors are considered benign in dogs. 13
Specific procedural measures must also be taken to avoid false-positive (clonal) results. All samples should be run in duplicate, along with both native and denatured forms; if this practice is not followed, pseudoclones can occur, resulting in false-positive (clonal) results. 58,88 Pseudoclones are characterized by 1 or 2 bands that are of different size or nonreproducible when run side by side. 58,88,141 It is important to inquire whether a laboratory uses these methods; if it does not, results should be interpreted with caution. Oligoclonal results can also be somewhat difficult to interpret. Oligoclonal samples have 3 to 5 reproducible bands in duplicate analysis, and up to 3 bands are considered most consistent with a neoplastic cell population. 88,141 Oligoclonality can occur in some cases of chronic inflammatory disease, early neoplasia, neoplasms involving more than a single clone, certain infections, and other types of neoplasia. 15,45,88,103,127 Thus, again, it is important to use PARR testing in conjunction with clinical findings, histopathology, and immunophenotyping.
Ultimately, despite a polyclonal PCR result, if histologic features and immunohistochemical results are consistent with lymphoma, a diagnosis of a suspected lymphoma should still be made, and additional biopsy samples obtained at a later time point should be tested with PARR. Similarly, in cases where histology and immunophenotyping are inconclusive, but PARR testing indicates a clonal proliferation of either B or T lymphocytes, continued patient follow-up is required to determine if the clonal result is predictive of the patient developing lymphoma. 4 As the characterization of both feline and canine immunoglobulin genes of B cells and TCR genes of T cells continues to improve, assays with improved sensitivity and specificity will continue to be developed but should still never be used as a stand-alone test.
Detection of c-Kit Mutations for Prognosis and Therapeutic Decision Making in Canine Neoplasms
While histopathological examination of biopsy samples is used routinely for tumor diagnosis, molecular analysis of biopsy samples may provide more precise prognostication and in-depth guidance for effective therapeutic options. One of the most widely used molecular tests for prognosticating neoplastic diseases in humans that also has remarkable clinical implications is the detection of c-Kit mutations by PCR. Somatic mutations in c-Kit have been associated with a number of neoplastic entities in humans, including gastrointestinal stromal tumors (GISTs), mast cell diseases, leukemia, myeloproliferative diseases, melanoma, and germ cell tumors. 17,40,65,73 Activating mutations of c-Kit have been identified in mast cell tumors (MCTs) and GISTs in dogs and cats as well (Fig. 6). Moreover, detection of mutations of c-Kit in these tumors has been associated with prognosis and targeted therapy.
The KIT protein, the product of the c-Kit proto-oncogene, is classified as a tyrosine kinase (TK) receptor subclass III. 16 Activation of KIT occurs through binding of the KIT ligand, stem cell factor (SCF), which is produced by fibroblasts and endothelial cells. Binding of SCF to KIT results in receptor dimerization and change in conformation of the receptor, leading to the stabilization of the receptor-receptor interaction. In nonneoplastic human and canine mast cells, KIT activation results in various cellular responses, including differentiation, proliferation, growth, survival, adhesion, and chemotaxis. 112 Gain-of-function mutations of c-Kit cause constitutional tyrosine phosphorylation and downstream activation independent of ligand binding. 84
Immunohistochemistry for KIT labeling is a standard test to differentiate canine GISTs from soft tissue sarcomas. GISTs are derived from the interstitial cells of Cajal that express KIT, while all other nonangiogenic nonlymphogenic intestinal sarcomas are negative for KIT. Because GISTs can exhibit a more aggressive biological behavior than nonangiogenic nonlymphogenic intestinal sarcomas, detection of KIT expression or a c-Kit mutation is necessary to accurately diagnose, prognosticate, and select proper therapeutic strategies. While canine mast cells normally express KIT, different KIT expression patterns have been reported in canine MCTs. 137 Nonetheless, although there is significant correlation between these KIT expression patterns and disease-free and overall survival times of affected dogs, KIT expression patterns alone cannot reliably predict the biological behavior of each individual canine MCT.
Activating mutations in c-Kit can easily be detected within tumor tissue by gel-based PCR using primers specific to exons that carry the mutation (Fig. 6C). Activating mutations have most commonly been reported in exons 8, 9, 11, and 17 (Fig. 6D). While the test can easily be performed on freshly collected tumor samples, including fine-needle aspirates, DNA from tumor samples can also be extracted from FFPE tissues. The main limitation of detecting c-Kit mutations by PCR are inadequate numbers of tumor cells and failure of proper extraction (as previously discussed). Selection of FFPE blocks that contain sufficient tumor tissue is crucial for accurate detection of mutations. If there are not enough neoplastic cells with mutations in the tumor sample, wild-type alleles from nonneoplastic cells could be amplified and result in a false-negative result.
Ordinarily, the juxtamembrane domain of KIT (encoded by exon 11) serves as a negative regulatory domain, which prevents kinase activation in the absence of ligand. Mutations in the juxtamembrane domain disrupt the negative regulation of the kinase domain and thereby result in uncontrolled receptor activation. 70 Mutations in the juxtamembrane domain have been reported in up to 33% of canine cutaneous MCTs 139,152 and in 35.3% of canine GISTs. 38 Interestingly, the DNA sequences of c-Kit mutations observed in canine GISTs are similar to mutations in human GISTs (Fig. 6D). Recently, a similar mutation has also been detected in a cat with a gastric GIST. 90
Another location for activating c-Kit mutations in canine and feline MCTs that cause receptor phosphorylation and activation without binding of ligand is in exon 8. In canine MCTs, mutations in exon 8 are far less common than mutations in exon 11 and reportedly occur in less than 3% of affected dogs. 67 In contrast to dogs, c-Kit mutations in feline MCTs have been identified only in exon 8 and have been proposed to play an important role in feline mast cell tumorigenesis. 48
In canine MCTs, there is significant association between c-Kit mutations in exon 11 and a high grade designation. 152 Dogs with MCTs harboring such a c-Kit mutation have been reported to have significantly shorter overall survival and disease-free survival times. Moreover, c-Kit mutations are also significantly associated with aberrant KIT protein localization. 138
In addition to KIT expression patterning that results in accurate tumor prognosis, detection of c-Kit mutation will provide the information necessary for predicting proper therapeutic response. Recent therapeutic breakthroughs in the treatment of KIT signaling-driven tumors involve the development of tyrosine kinase inhibitors (TKIs). TKIs are promising new agents for selective, specific inhibition of malignant cell growth, promotion of apoptosis, and inhibition of angiogenesis and metastasis of tyrosine kinase receptor signaling–mediated tumors. Canine MCTs with activating mutations in exon 11 respond well to TKIs that are now readily available for dogs. 42,47,61,69,111 Furthermore, high-grade MCTs lacking mutations or aberrant KIT expression have been reported to respond better to a chemotherapy protocol composed of vinblastine and prednisone than to TKI. 140 Similar to the results in dogs, cats with MCTs harboring a mutation in exon 8 have also been reported to have a favorable response to TKIs. 41,48 In conclusion, molecular analysis using PCR to detect c-Kit mutations in FFPE tumor samples allows more accurate prediction of clinical behavior, determination of the risk of metastases, and assistance in determining appropriate treatment strategies for KIT signaling-driven tumors.
Localization of Nucleic Acid Within Microscopic Lesions
In Situ Hybridization
To establish the cause or pathogenesis of certain diseases, it is often necessary to determine the presence of infectious agents or abnormal gene expression in association with morphologic changes within tissues. While PCR is a highly sensitive tool and often considered the gold standard for detection of an infectious agent in the tissues, it does not allow for the correlation of the microscopic lesions directly with the causative agent. This can be crucial because tissues may harbor vaccine, latent virus, or otherwise clinically asymptomatic virus that could potentially be misinterpreted as the cause of disease. A good example is asymptomatic porcine circovirus type 2 (PCV-2) infection in pigs, which is much more common than actual disease caused by this virus. Therefore, demonstrating PCV-2 infection via PCR is not sufficient for the diagnosis of PCV-2–associated diseases. Diagnosis requires localization of the virus within characteristic lesions (Fig. 7). 117
IHC is presently the most frequently used molecular tool for the detection of a specific protein target within microscopic lesions of FFPE tissue. Some of the main disadvantages of IHC are the limited availability of species-specific antibodies and problems with the continued supply and batch variation of some antibodies. 33 Another issue is the specificity of some antibodies, 33,114 mostly with regard to infectious agents.
In situ hybridization (ISH) is another powerful molecular tool that allows detection of a particular DNA or mRNA sequence within cells and tissue sections. In essence, any nucleic acid sequence can be specifically detected by the use of a probe that is the “antisense” or reverse complementary sequence of the target.
ISH can be used to identify and localize nucleic acid of infectious agents within lesions, as well as localize genes, their transcripts, and chromosomal aberrations. 11,27,114 Any nucleic acid sequence can specifically be detected by the use of either radioactively or chemically labeled DNA or RNA probes. Radioactive probes are the most sensitive. 142 Nonetheless, nonradioactive probes, generally labeled with digoxigenin, biotin, direct labeling dinitrophenol (DNP), or fluorescence, produce quicker results and are safer to handle. Biotinylated probes work well but may present high levels of background because of endogenous biotin within tissues. 142 Fluorescent-labeled probes are especially useful for the direct examination of microbiological or cytological specimens using confocal microscopy. 142 Digoxigenin or DNP-labeled probes have the advantage that their antibody detection has no cross-reactivity with animal tissues and that they can be visualized using a chromogen such as nitroblue-tetrazolium (NBT), similar to IHC. With digoxigenin or DNP-labeled oligonucleotide probes, the synthesis is easy to achieve through automated manufacturing and end labeling. 3,71 High-performance liquid chromatography (HPLC) purification of the labeled probe is highly desirable to increase probe sensitivity by removing nonlabeled probe. These types of probes are also more stable and are easier to work with because of their resistance to degradation by the ubiquitous, contaminating RNAse. 71 Nonetheless, due to lower labeling efficiency and decreased target size, oligonucleotides are less sensitive than, for instance, RNA probes (riboprobes). Such sensitivity issues can be overcome by designing oligonucleotide probes for a repeated, accumulated target within the tissue or cell. Sensitivity may also be increased by using a mixture of multiple oligonucleotide probes or degenerate probes. 123 In our experience using nonamplified detection systems, digoxigenin or DNP-labeled oligoprobes are usually limited to detect target concentrations that would be recognized by up to 25 cycle thresholds by a complementary PCR reaction.
Historically, ISH is regarded as a time-consuming, labor-intensive, and technically complex method, the optimization of which can be difficult. A proposed rational design of oligonucleotide probes that takes into account melting temperature, runs of identical nucleotides, guanine-cytosine (GC) content, length, and secondary structures allows for a standard ISH protocol that keeps constant basic parameters such as tissue fixation, hybridization conditions, and washing procedures. 31 This approach has worked well in our laboratory and represents another great advantage of oligonucleotide probes compared with riboprobes. Automation of the ISH procedure has further improved consistency, reliability, and feasibility of this assay, with increased throughput capability. 95
Probes for infectious agents can either be generic or species specific. By screening tissues first with a generic probe (eg, papillomavirus, Leishmania, Chlamydophila), it is possible to search for a wider range of agents, followed by the use of species-specific probes at a later stage to speciate the agent of interest (eg, Leishmania infantum, Chlamydophila psittaci). 82 ISH can also be used to localize mRNA and to determine expression levels of specific transcripts such as oncogenes. 27 This allows for information on the distribution and expression pattern of the target within tissues, providing spatial and temporal information about infectious agents and genes as well as information on disease progression and levels of cell differentiation and functional activity. 27
Careful selection of the proper test is crucial for successful identification of the target. For instance, papillomaviral mRNA codes for structural proteins that lead to accumulation of a large number of virions in the stratum granulosum and corneum. Thus, nonkeratinizing epithelial neoplasms associated with papillomavirus infection may be negative with IHC for this virus since commercial antibodies target viral proteins that are produced only in the late stages of viral replication. 91 Detection of papillomavirus within equine and feline sarcoids will similarly require testing for the viral nucleic acid rather than for viral proteins. ISH can also differentiate sites of productive versus nonproductive viral infection when using either multiple probes that target mRNA and DNA or probes in combination with IHC (Fig. 8). 57 In addition, through dual ISH/IHC labeling, cells that are actively producing virus may be able to be distinguished from those solely involved in phagocytosis or uptake of the target. 102 Depending on the probe design, ISH can also be used to exclusively detect the replicative form of a virus. 102 Similarly, combining ISH and IHC is useful to identify, respectively, which cells are producing and which cells are the targets of secreted proteins such as cytokines. 116
The major pitfalls with both IHC and ISH relate to the differentiation between specific and nonspecific or background labeling. Some probes will bind nonspecifically to neurons, glandular epithelial cells, and collagen. 11 Likewise, certain immunohistochemical detection methods will label endogenous enzymes or other molecules such as biotin. 14,66,105 Besides the specificity and sensitivity of the antibodies or probe, other variables that can affect the labeling results include tissue processing, retrieval methods, and the detection system. 14,24,66,105,106 For instance, secondary polyclonal antibodies may cross-react with pathogens that are morphologically similar to the target, with the potential for incorrect interpretation. Toxoplasma gondii cysts and tachyzoites in tissues cross-reacted with a rabbit polyclonal anti–digoxigenin antibody in ISH for Leishmania, probably because the antiserum was raised in an animal subclinically infected with T. gondii. This problem, which could easily lead to false-positive diagnosis considering the similar morphology between Leishmania and T. gondii, was solved by replacing the polyclonal antibody with a monoclonal anti–digoxigenin antibody. 82 Differences in labeling intensity and variable assessment of the degree of positivity, mainly in the case of some nuclear antigens such as Ki-67, p53, and estrogen receptor, may also lead to significant interobserver variation. 66 Knowledge of the expected labeling pattern within the tissue and subcellular compartment is therefore essential for correct interpretation, along with the need for proper controls to ensure accurate results. The ideal control for IHC and ISH contains the desired target in a detectable concentration and is processed in a similar way as the tissue sample(s) to be tested. Pellets of cultured cells that either were infected with the infectious agent of interest or overexpress the target gene can fulfill these criteria well. After resuspension in HistoGel (Richard-Allan Scientific, Kalamazoo, MI), such cell pellets can then be fixed in 10% NBF and routinely processed for histopathology, IHC, and ISH like any other tissue sample. 53
Other Methods to Detect Intralesional Targets
Significant advancement has been made in recent years in the development of both molecular-based diagnostic techniques and molecular-targeted therapies. Molecular profiling of diseases has profited immensely from microdissection techniques, which enable investigators to evaluate characteristics of specific cell populations. Laser-capture microdissection, in particular, allows for isolation of specific cell types from FFPE tissues and subsequent nucleic acid and protein analysis through standard laboratory methodologies. 18,50 In addition, protein profiling (proteomic) assays, such as microarrays, Raman spectrometry, and matrix-assisted laser desorption ionization (MALDI) imaging mass spectrometry, are being adapted and perfected for FFPE specimens to determine and validate new biomarkers, as well as to further elucidate disease pathogenesis. More detailed information on these techniques and their application on FFPE tissue is available in reviews within this issue 148 and elsewhere. 78,104
Conclusions
A review of existing data clearly shows that formalin fixation has a negative effect on nucleic acids. Therefore, it is important for a laboratory performing molecular assays on FFPE tissues to implement and adhere to protocols that minimize nucleic acid damage. The process of nucleic acid preservation should start with minimizing the interval between tissue collection and fixation. The formalin buffering system, tissue size, length of fixation, and the embedding procedure can subsequently all affect nucleic acid quality.
DNA extraction from FFPE tissue is in general well established, taking into account that the upper size limit of the extracted DNA is approximately 300 to 400 bp with most extraction protocols. RNA extraction is much more challenging, and the consistently extracted maximum size is typically 100 to 200 bp. It is very encouraging that commercial kits, especially those that are specifically marketed for use on FFPE tissue, are more efficient for use in FFPE tissues than the equivalent kits that are routinely used on unfixed tissue. Downstream nucleic acid detection assays can be either morphological or liquid phase based. On the basis of preferred amplicon size and assay efficiency, we consider probe-based or fluorescent dye–based real-time PCR to be the most suitable liquid phase detection system for current use on FFPE tissue extracts.
Most important, one diagnostic test is unlikely to provide a complete picture of the disease. A combination of selecting the proper tissue samples and applying the appropriate molecular tests allows for better understanding of the pathogenesis, identification of the target cells, monitoring of disease progression, and determination of the most appropriate patient management. The use of FFPE material in this setting is fundamental because this may be the only material available, in addition to enabling retrospective analyses using archived material. In modern diagnostic settings, communication between the different laboratory sections, including both the molecular biologists and the pathologists, is crucial.
Footnotes
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.