
Abstract
Neosporosis and toxoplasmosis are major causes of abortion in livestock worldwide, leading to substantial economic losses. Detection tools are fundamental to the diagnosis and management of those diseases. Current immunohistochemistry (IHC) tests, using sera raised against whole parasite lysates, have not been able to distinguish between Toxoplasma gondii and Neospora caninum. We used T. gondii and N. caninum recombinant proteins, expressed in Escherichia coli and purified using insoluble conditions, to produce specific polyclonal rabbit antisera. We aimed to develop species-specific sera that could be used in IHC on formalin-fixed, paraffin-embedded (FFPE) tissue sections to improve the diagnosis of ruminant abortions caused by protozoa. Two polyclonal rabbit sera, raised against recombinant proteins, anti–Neospora-rNcSRS2 and anti–Toxoplasma-rTgSRS2, had specificity for the parasite they were raised against. We tested the specificity for each polyclonal serum using FFPE tissue sections known to be infected with T. gondii and N. caninum. The anti–Neospora-rNcSRS2 serum labeled specifically only N. caninum–infected tissue blocks, and the anti–Toxoplasma-rTgSRS2 serum was specific to only T. gondii–infected tissues. Moreover, tissues from 52 cattle and 19 sheep previously diagnosed by lesion profiles were tested using IHC with our polyclonal sera and PCR. The overall agreement between IHC and PCR was 90.1% for both polyclonal anti-rNcSRS2 and anti-rTgSRS2 sera. The polyclonal antisera were specific and allowed visual confirmation of protozoan parasites by IHC, but they were not as sensitive as PCR testing.
Keywords
Neospora caninum and Toxoplasma gondii are apicomplexan intracellular protozoan parasites. 26 N. caninum is known to be a major cause of endemic and epidemic abortions in cattle around the world and has been shown to seriously impact the economic performance of the dairy and beef industries. 26 N. caninum is mainly responsible for abortions in cattle, but it has also caused characteristic lesions in small ruminants with clinical signs and lesions similar to those induced by T. gondii. 30
Toxoplasmosis is well known for causing abortions in sheep, and T. gondii can also be found in other ruminants, including cattle.3,16,28 Furthermore, toxoplasmosis is a major zoonosis; in pregnant women, T. gondii can infect the fetus and cause miscarriage or result in congenital toxoplasmosis, which can cause brain damage. In immunocompromised people, T. gondii infection can be very severe and even life-threatening. 20 N. caninum and T. gondii cannot be distinguished solely by clinical and microscopic findings, given that they share many common biologic and morphologic similarities. 32 The detection of protozoan parasites is difficult even in tissues from clinically infected animals because parasites are not always associated with lesions.9,26 Etiologic detection tests such as immunohistochemistry (IHC) and PCR are required to establish the etiologic cause of abortion and thus permit the design of effective control strategies.
IHC can identify one or more immunogenic epitopes, allowing visualization and establishment of the distribution of the pathogen within tissue sections.14,35 N. caninum polyclonal antiserum raised against whole parasite lysates can cross-react with other cyst-forming parasites, such as T. gondii and Sarcocystis spp. 27 Antibodies developed using whole parasite lysates often cross-react with other protozoa and hence are unsuitable for distinguishing between these closely related parasites using IHC.13,17 Various studies have shown that using specific protozoan proteins in their native and recombinant forms results in reduced cross-reactivity with other coccidian parasites. 35
Formalin-fixed, paraffin-embedded (FFPE) tissue sections have been used most widely for clinical diagnosis. However, for FFPE tissue sections, it is known that formaldehyde cross-links proteins, which results in loss of antigenicity as a result of the formation of methylene bridges, making binding of specific antibodies difficult during IHC. 28 Antigen retrieval (AR) methods allow specific recognition of proteins from FFPE tissues by breaking these bridges and exposing the antigenic sites to allow antibodies to bind. 31
Similarly for PCR analysis, when fresh tissues are not available, FFPE tissue samples can be used for the identification of parasite DNA.2,33 However, the extraction of DNA from FFPE samples remains challenging given that various fixatives, including formaldehyde, cause cross-linkage. The cross-linkage of DNA by formaldehyde can cause fragmentation, strand breaks, and chemical modifications, which can inhibit PCR. 31 Fixation decreases PCR sensitivity, thus making PCR amplification of large- and high-molecular-weight DNA fragments considerably more difficult.12,22 Nevertheless, DNA can be successfully purified, and the amplification of fragments of <500 bp via PCR is possible. 25 Various PCR assays have been described that can detect N. caninum, T. gondii, and Sarcocystis spp., targeting multicopy rDNA, containing the 18S ribosomal RNA gene and the internal transcribed spacer 1 (ITS1) region, from frozen tissue samples. 10 Several commercialized protocols allow the amplification of protozoan parasite DNA from FFPE tissue samples.5,34
We aimed to produce species-specific antibodies, raised against recombinant proteins of N. caninum and T. gondii, which could then be used to detect and characterize protozoan parasites in diagnostic and research studies investigating cases of abortion in ruminants. We also aimed to evaluate the diagnostic sensitivity and specificity of the species-specific antisera in IHC in direct comparison to PCR results conducted on DNA extracted from matching FFPE tissue sections in which we detected protozoan parasites and determined their species.
Materials and methods
Recombinant protein production and testing
N. caninum (NcSRS2) and T. gondii (TgSRS2) surface antigen genes were selected and used to produce recombinant proteins. The chosen regions were polymorphic and amplified using gene-specific primers (Table 1). DNA samples, extracted from N. caninum (Nc1) and T. gondii (M4), were used as positive controls; negative water was included as negative controls in all PCRs. Positive amplicons were sequenced using the MWG Eurofins (Ebersberg, Germany; Table 1). DNA sequences were analyzed using BLAST (https://blast.ncbi.nlm.nih.gov/Blast.cgi) to confirm the species and to determine sequence identity.
Cloning details for the generation of recombinant proteins from Neospora caninum and Toxoplasma gondii.

Lowercase nucleotides represent the restriction enzyme sites; capitalized nucleotides represent the actual primer.
Each PCR reaction contained 2 µL of 10× custom PCR mix, 13.8 µL of water, and 2 µL of sample DNA using PCR conditions described previously,4,24 with the exception of an annealing temperature of 60°C for 1 min. Positive PCR amplicons were purified and cloned. 24 Plasmids were verified by DNA sequencing, and DNA inserts were digested, purified, and ligated into pre-digested expression vector pQE-30/31 (QIAexpressionist; Qiagen) using appropriate restriction enzymes (Table 1). Expression constructs were verified by sequencing and used to express the recombinant proteins in Escherichia coli M15 (pREP4) according to the manufacturer’s instructions. The proteins were purified under denaturing conditions (HisPur Ni-NTA spin columns; Thermo Fisher) according to the manufacturer’s instructions, except for using 6 wash steps with 2 resin-bed volumes of denaturing stock wash buffers: 20 mM NaH2PO4, 300 mM NaCl, 25 mM imidazole, 8 M urea and 20 mM NaH2PO4, 300 mM NaCl, 50 mM imidazole, 8 M urea. Recombinant proteins were dialyzed into 2 M urea (Slide-A-Lyzer G2 dialysis cassette; Thermo Fisher) and concentrated (Vivaspin 6 at 10,000 MWCO; GE Healthcare) for 15 min. Protein concentrations were determined (Pierce BCA protein assay kit; Thermo Fisher).
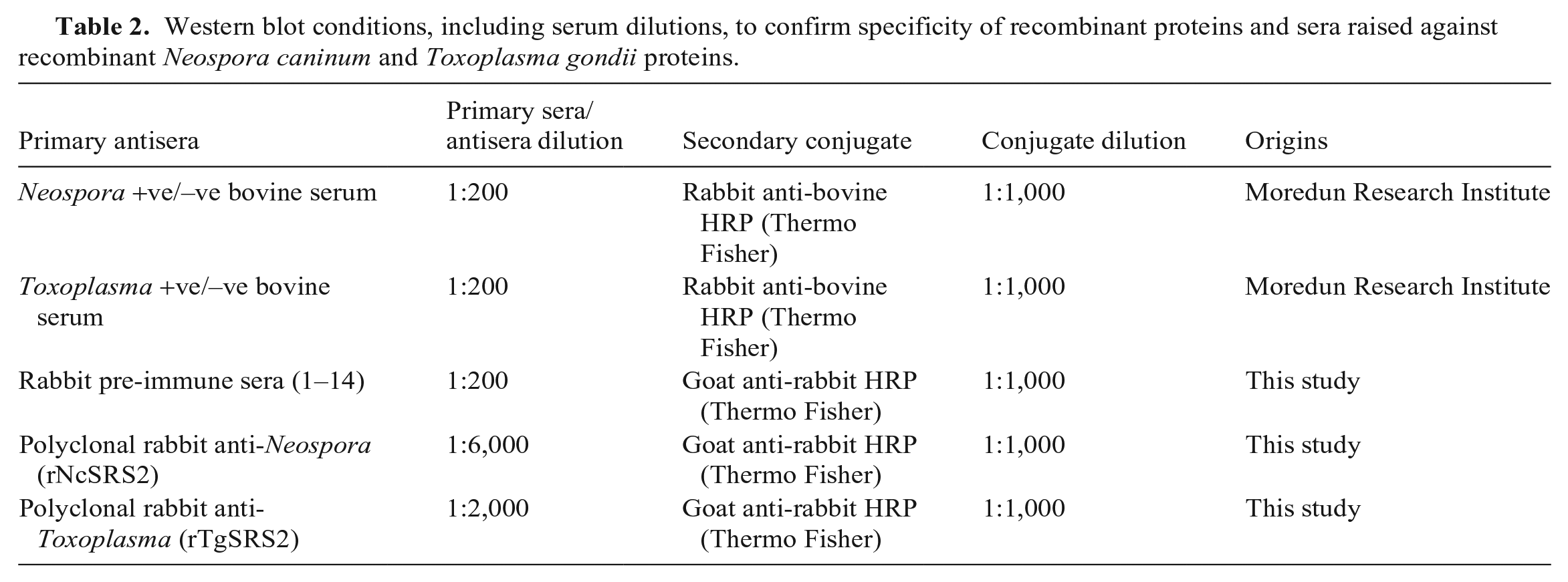
Western blots were performed to identify reactivity and cross-reactivity among the N. caninum and T. gondii recombinant proteins with positive and negative bovine antisera (Table 2). 35 Briefly, a recombinant protein concentration of 3 ng/mL was used. We used 10 µL of positive controls of N. caninum water-soluble antigen fraction (NAF; 25 μg/mL) and T. gondii water-soluble antigen fraction (TAF; 27 μg/mL). The proteins were transferred to a nitrocellulose blotting membrane (Amersham Protran premium 0.45 μm, GE Healthcare; XCell IITM blot module, Invitrogen) according to the manufacturers’ instructions and incubated overnight at 4°C in transfer buffer (1× Novex Tris-glycine; Thermo Fisher). Membranes were incubated in 4% non-fat dried milk (Marvel) for 1 h with primary antisera (Table 2). Membranes were washed 3 times for 5 min after each incubation with a wash buffer (0.05% Tween 80, 500 mM NaCl, 1× PBS). The secondary conjugate was incubated for 1 h (Table 2). Protein bands were detected (SuperSignal west pica chemiluminescent substrate kit; Thermo Fisher) and visualized (ImageQuant Las-4000 multi-mode imager; GE).
Western blot conditions, including serum dilutions, to confirm specificity of recombinant proteins and sera raised against recombinant Neospora caninum and Toxoplasma gondii proteins.
Production of rabbit polyclonal sera
Our study was carried out in strict accordance with the Animals (Scientific Procedures) Act 1986 (https://www.legislation.gov.uk/ukpga/1986/14/contents) and in compliance with all UK Home Office Inspectorate regulations under PPL70/8627.
The pre-immune sera from 14 rabbits (Orgyen Antibodies) were tested to rule out nonspecific reactivity with N. caninum or T. gondii. Western blot analysis was performed using 10 µL of rNcSRS2 and rTgSRS2 recombinant proteins (3 ng/mL) and NAF/TAF (22.5 μg/mL; Table 2). IHC was carried out using the rabbit pre-immune sera on FFPE positive control blocks from the pathology archive at the Moredun Research Institute (Midlothian, Scotland, UK), which were previously confirmed to be N. caninum–positive (4 bovine brain and heart tissue blocks; 2 T. gondii–positive ovine brain tissue blocks). The presence of protozoan parasites had been confirmed previously by lesion profile and the presence of parasite on H&E staining and IHC.
Four-µm thick paraffin-embedded tissue sections were cut and placed onto glass slides (SuperFrost Plus; Thermo Scientific). Tissues were dewaxed in xylene for 20 min and rehydrated through graded ethanol. The endogenous peroxidase was quenched by immersion of the slides in 3% hydrogen peroxide in methanol solution for 20 min. No AR method was used for testing rabbit pre-immune sera. Slides were assembled with cover plates in Sequenza racks (Thermo Scientific) and washed 3 times with Tris-buffer saline (TBS; 1 M Tris HCl, 5 M NaCl, pH 7.6) until the wash buffer was fully drained following each incubation. Tissues were first blocked with 25% normal goat serum in TBS. Slides were next incubated with rabbit pre-immune sera overnight at 4°C. Antigen-antibody reactions were detected using a horseradish peroxidase–labeled polymer. The slides were treated with peroxidase substrate solution (AEC; Vector) following the manufacturer’s instructions, and counterstained with Mayer hematoxylin and Scot tap water substitute for 2 min each. Slides were mounted with a coverslip (ImmunoHistoMount, Sigma-Aldrich; permanent Consul Mount, Shandon).
Polyclonal sera were produced by Orgyen Antibodies by using rNcSRS2 and rTgSRS2 to immunize rabbits whose pre-immune sera had no reactivity with protozoan parasites by western blots and IHC. New Zealand white rabbits were immunized 4 times (weeks 1, 4, 8, 12) with 150 μg per dose of each recombinant protein. Per injection, 0.25 mL of recombinant proteins at stock concentrations of 600 µg/mL were emulsified with Freund adjuvant and administered into each rabbit. Test bleeds were taken during weeks 5 and 9. Rabbits were exsanguinated at week 13.
Standardization and specificity of rabbit polyclonal sera
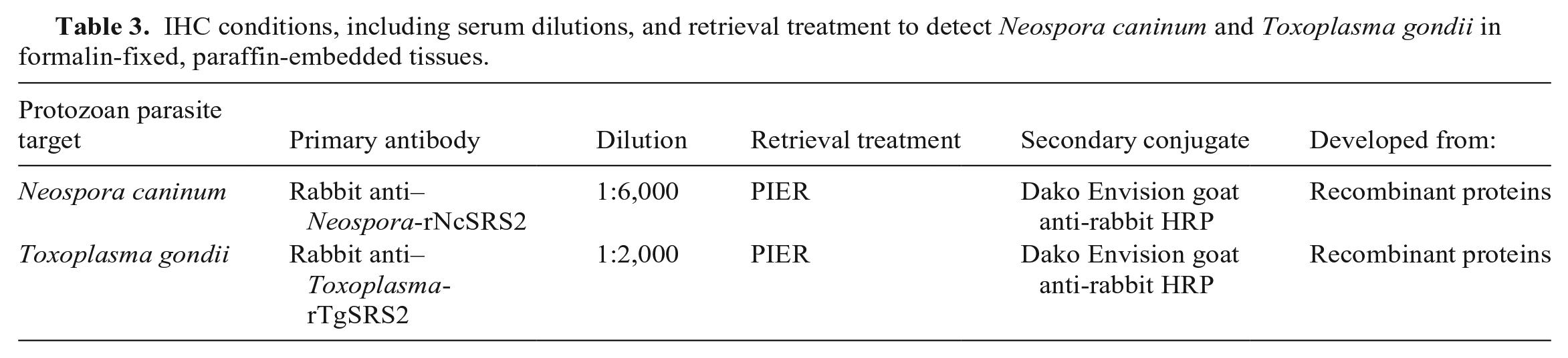
Preliminary unpublished studies, conducted at the Moredun Research Institute, demonstrated that polyclonal sera raised against whole tachyzoite lysates were cross-reactive with N. caninum, T. gondii, and Sarcocystis spp. by IHC. The reactivity of each polyclonal antiserum raised against the recombinant proteins was evaluated by IHC on FFPE tissues using the same method as described above under specific conditions. To establish optimal working conditions, dilutions (1:100; 1:200; 1:500; 1:1,000; 1:2,000; 1:3,000; 1:4,000; 1:6,000; 1:8,000; 1:10,000) were tested for each antiserum. Different AR methods were used to “de-mask” the modification of antigen targets caused by fixation. The optimal AR methods were evaluated by using: no AR treatment, heat-induced epitope AR (HIER; citrate buffer, 10 mM citric acid, at pH 6.0 in a pre-heated autoclave at 121°C for 10 min), and protease-induced epitope AR (PIER; 0.1% protease, Streptomyces griseus [Sigma-Aldrich] in TBS at 37°C for 10 min). The method with the highest labeling intensity and optimal working conditions was the PIER method and a dilution of 1:6,000 for the serum raised against rSRS2 from N. caninum, and serum raised against rSRS2 from T. gondii at a dilution of 1:2,000 (Table 3).
IHC conditions, including serum dilutions, and retrieval treatment to detect Neospora caninum and Toxoplasma gondii in formalin-fixed, paraffin-embedded tissues.
Cross-reactivity of rabbit anti-rTgSRS2 and rabbit anti-rNcSRS2 was tested using western blot and IHC analysis. Western blots were performed as described above with the exception of using the polyclonal sera (Table 2). IHC analyses were performed, using the PIER method, on 2 positive Neospora (canine brain), 2 positive Toxoplasma (1 murine liver and 1 feline brain), and 2 positive Sarcocystis (ovine heart) control tissue sections. Rabbit pre-immune sera were used as negative antibody controls.
Protozoan detection on archived FFPE tissue blocks by IHC and PCR
The polyclonal antisera (anti-rNcSRS2, anti-rTgSRS2) were tested using the optimized conditions (Table 3) on a selection of FFPE tissue blocks obtained from the Moredun pathology and surveillance archive. These tissue blocks were from 71 animals (52 cattle, 19 sheep) and included fetal brain, placenta, and skeletal muscle with lesions compatible with protozoan infection. One block per animal was selected, and one 4-µm thick paraffin-embedded tissue section was cut and used per antiserum.
For PCR analysis, two 20-µm thick FFPE tissue sections were cut from the same blocks to confirm the presence of DNA from protozoan parasites. DNA was extracted (QIAamp DNA FFPE tissue kit, 56404; Qiagen) according to the manufacturer’s instructions, with the following modifications. Sections were de-paraffinized with xylene and rehydrated in 100% ethanol. Approximately 300 µL of buffer ATL and 30 µL proteinase K (both supplied with the QIAamp DNA FFPE tissue kit) were used. Samples were lysed overnight at 56°C in a water bath. To increase DNA concentrations, columns were incubated for 5 min with 50 µL of ATE buffer (QIAamp DNA FFPE tissue kit) before centrifugation. The nucleic acid concentration and purity were determined (Nanodrop 1000 spectrophotometer; Thermo Fisher). Parasite DNA was detected using nested PCRs, and reaction conditions for the first- and second-round PCR were carried out as described previously (Table 4), 24 with the exception of the annealing temperature of 58°C for the 18S and 55°C for the ITS1 PCR assays in the second-round PCR assay. The first-round PCR assay used external primers that recognized apicomplexan parasites, including N. caninum, T. gondii, Sarcocystis spp., and Hammondia spp. (H. hammondi, H. heydorni, H triffittae), and the second-round PCR assay included the genus- and species-specific primers to distinguish among N. caninum, T. gondii, and Sarcocystis spp. DNA samples were tested in triplicate by PCR. N. caninum– and T. gondii–positive DNA samples were included as positive controls, and water was included as a negative control in the PCRs. Positive amplicons were sequenced (DNA sequencing service; MWG) using internal primers (NTS-18S-F1 and R1; Table 4). Sequences were analyzed using SeqMan Pro (DNASTAR) and BLAST searches to determine the species identified against published sequences.
Sequences of primers used for the detection of Neospora, Toxoplasma, and Sarcocystis spp. DNA from 18S rDNA and the ITS1 region.

Data analysis
We analyzed our data with R for statistical computing (v.4.0.2; https://www.r-project.org/). The proportion of positive samples for each test and parasite species were calculated, including 95% CIs based on a binomial probability distribution. 7 In the absence of a gold standard, agreement statistics between the IHC and PCR test results were computed based on combined data from both ovine and bovine species, namely, overall percent agreement, and associated 95% CI, along with Cohen kappa coefficient. 21 This latter measure ranges in [0; 1], with 0 meaning that the agreement is not better than random, and 1 meaning perfect agreement between tests. Statistical significance was concluded at the 5% significance level (p ≤ 0.05).
Results
Development, purification, and testing of recombinant proteins in western blots
Amplicons for the polymorphic regions of the NcSRS2 (1,057 bp) and TgSRS2 (540 bp) were generated and cloned into pQE expression vectors. Sequencing the expression vectors confirmed the presence of the desired open-reading frames, and >99% sequence identity to the published NcSRS2 and TgSRS2 sequences encoding predicted amino acid sequences of 354 for rNcSRS2 and 180 for rTgSRS2. All recombinant proteins were successfully purified, resulting in a rNcSRS2 protein with a predicted size of 37.2 kDa and a rTgSRS2 protein with a predicted size of 19.2 kDa. In both cases, the purified proteins were slightly larger on the SDS-PAGE gels, and for rTgSRS2, larger bands of approximately twice the size are visible, which represents protein dimers, and multimers that are even larger (Suppl. Fig. 1A, 1B).
Western blot results revealed that Neospora-positive bovine sera reacted with the rNcSRS2 and NAF; no reaction was seen with rTgSRS2 and TAF (Suppl. Fig. 2A). No reactions were observed with recombinant proteins or TAF with Neospora-negative bovine sera; a very weak reaction was seen with NAF (Suppl. Fig. 2B). Furthermore, western blot results revealed that the Toxoplasma-positive bovine sera reacted with the rTgSRS2 and TAF; no reaction was seen with rNcSRS2 and NAF (Suppl. Fig. 2C). No reactions were observed with recombinant proteins and NAF with Toxoplasma-negative bovine sera (Suppl. Fig. 2D). However, a slight reaction with TAF was observed with Toxoplasma-negative bovine sera (Suppl. Fig. 2D).
Selection of rabbits for immunization
Both the western blots and IHC were performed to test the reactivity of the rabbit pre-immune sera with NAF/TAF and recombinant proteins. For the western blot analysis, 4 of 14 pre-immune sera reacted with recombinant proteins rNcSRS2 and rTgSRS2, and none of the sera reacted with NAF/TAF. The pre-immune sera screen via IHC showed strong background and nonspecific binding of 11 of 14 sera with N. caninum and T. gondii. Based on these combined results, 3 rabbits had unreactive sera, from which 2 were used for antibody production.
Optimization of rabbit polyclonal sera
IHC titration showed that rabbit anti-rNcSRS2 and rabbit anti-rTgSRS2 had the highest signal:background ratio at a dilution of 1:6,000 for N. caninum and 1:2,000 for T. gondii. In the absence of a retrieval method, parasites had very faint labeling for both polyclonal sera (Suppl. Fig. 3A, 3D). Parasites had positive labeling when treated with HIER, but histologic quality was altered (Suppl. Fig. 3B, 3E). Slides treated with PIER had the best labeling of the protozoan parasites (Suppl. Fig. 3C, 3F). No labeling was observed with the rabbit pre-immune sera.
Rabbit polyclonal antisera specificity
Western blot results revealed that rabbit anti-rNcSRS2 reacted with rNcSRS2 and NAF, and did not react with TAF or rTgSRS2 (Suppl. Fig. 4A). Rabbit anti-rTgSRS2 had strong reactivity with rTgSRS2, and no reactivity with NAF, TAF, or rNcSRS2 (Suppl. Fig. 4B). Antisera raised against rNcSRS2 and rTgSRS2 reacted with dimers and multimers of the purified recombinant proteins that they were raised against (Suppl. Fig. 4).
The rabbit anti-rNcSRS2 serum had specific labeling of N. caninum parasites using IHC (Fig. 1A), but no specific labeling of T. gondii (Fig. 1B). The rabbit anti-rTgSRS2 serum had specific labeling of T. gondii parasites using IHC (Fig. 1C), but no specific labeling of N. caninum (Fig. 1D). No labeling was observed on the Sarcocystis tissue control (data not shown).
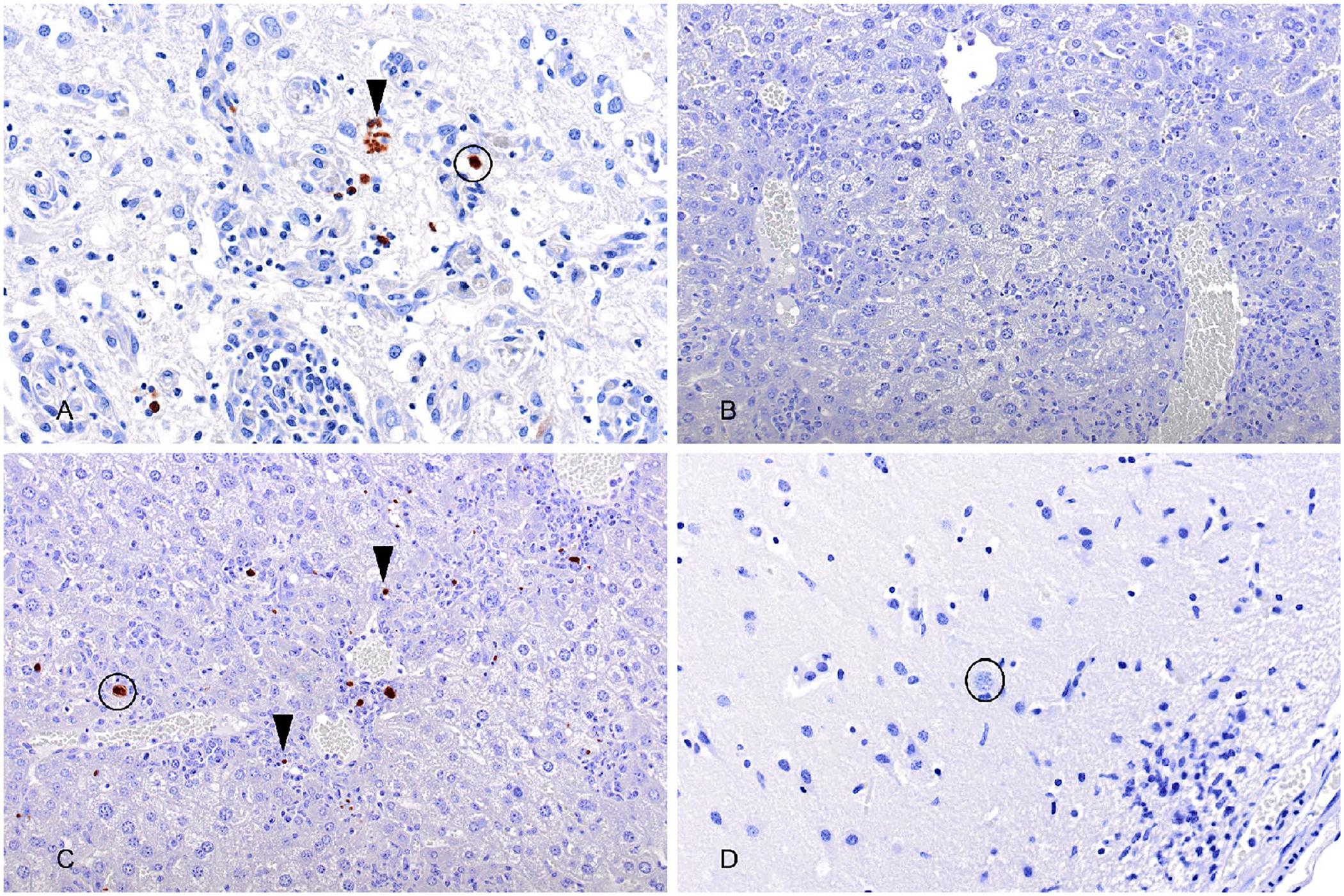
Immunohistochemistry with anti-rNcSRS2 or anti-rTgSRS2 antisera on Neospora caninum– or Toxoplasma gondii–positive control tissues.
Protozoan detection on archived FFPE tissue blocks by IHC and PCR
A total of 71 ruminant samples were tested by IHC and PCR in parallel to detect infections by protozoan parasites (Suppl. Fig. 5). IHC using rabbit anti-rNcSRS2 serum showed that 10 of 71 (14.1%; 95% CI: 7.0–24.4%) ruminant tissue samples were positive (Fig. 2). A total of 10 of 52 (19.2%; 95% CI: 9.6–32.5%) bovine samples had positive labeling for N. caninum (Fig. 2). No ovine samples were positive for N. caninum (Fig. 2).
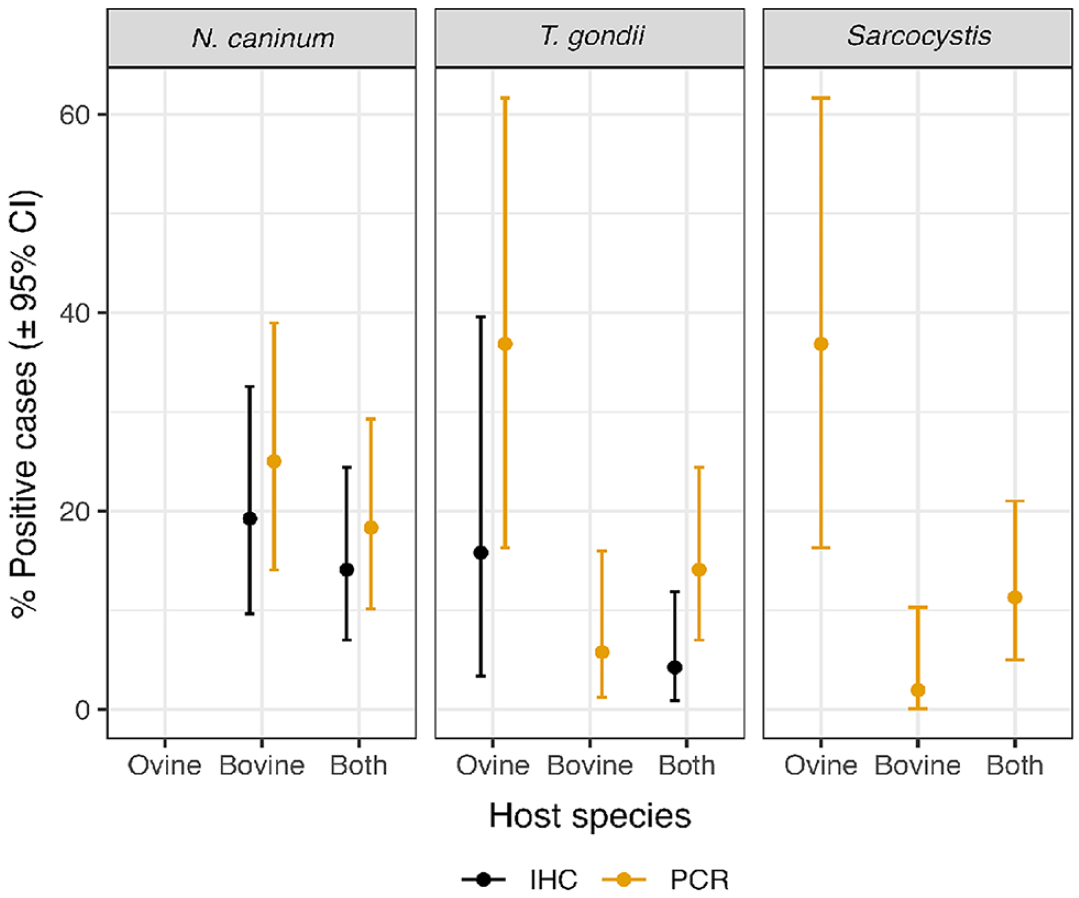
Summary of positive Neospora caninum and Toxoplasma gondii results from the IHC and PCR tests, and Sarcocystis spp. results from the PCR tests, both for ovine and bovine samples separately and for all ruminants combined. The percentage positive column shows the percentage of positive cases and the lower and upper confidence limits (LCL, UCL, respectively) columns show the estimated LCLs and UCLs of a 95% CI for this percentage based on a binomial probability distribution.
PCR results revealed that 13 of 71 (18.3%; 95% CI: 10.1–29.3%) ruminant samples were positive for N. caninum; 13 of 52 cattle samples (25.0%; 95% CI: 14.0–38.9%) were positive for N. caninum (Fig. 2). No ovine samples were positive by PCR for N. caninum (Fig. 2).
IHC using rabbit anti-rTgSRS2 serum showed labeling in 3 of 71 ruminant tissue samples (4.2%, 95% CI: 0.9–11.9%; Fig. 2); 3 of 19 (15.8%; 95% CI: 3.4–39.6%) ovine samples showed positive labeling for T. gondii, yet no bovine samples were positive for T. gondii (Fig. 2).
Using T. gondii–specific PCR, 10 of 71 (14.1%; 95% CI: 7.0–24.4%) ruminant samples were positive, of which 3 of 52 (5.8%; 95% CI: 1.2–15.9%) were cattle samples and 7 of 19 (36.8%; 95% CI: 16.3–61.6%) were ovine samples (Fig. 2).
PCR analysis revealed that 8 of 71 (11.3%; 95% CI: 5.0–21.0%) ruminant samples were positive (at least one of the triplicate PCRs) for Sarcocystis spp., 1 of 52 (1.9%; 95% CI: 0.1–10.3%) cattle samples was positive for S. cruzi, and 7 of 19 ovine samples (36.8%; 95% CI: 16.3–61.6%) were positive for Sarcocystis species (Fig. 2). Sarcocystis tenella was detected in 3 animals and S. gigantea in 4 according to BLAST results with >99% sequence identity (S. tenella L24383; S. gigantea L24384). One ovine placenta sample was positive for both S. tenella (by PCR) and T. gondii (by PCR and IHC), and one bovine brain sample was positive for both S. cruzi (by PCR) and N. caninum (by PCR and IHC; Suppl. Fig. 5).
When assessing all ruminant samples for N. caninum, 80% of IHC positive samples were also positive by PCR, whereas 8.19% of samples positive by PCR were negative by IHC. The overall agreement (including negative results) for N. caninum was 90.1% (95% CI: 82.0–94.8%), with a kappa statistic of 0.64 (p < 0.001). In the case of T. gondii, all IHC positive samples were also positive by PCR, whereas 10.3% of samples positive by PCR were negative by IHC. The overall percent agreement for T. gondii was 90.1% (95% CI: 84.4–93.9%), with a kappa of 0.42 (p < 0.001).
Discussion
Our specific polyclonal sera for N. caninum and T. gondii did not cross-react with the other pathogen and did not label Sarcocystis spp., demonstrating that specific polyclonal antisera can be applied to IHC testing of FFPE tissue sections for support of species-specific diagnosis of T. gondii– or N. caninum–induced abortions. Previous tests that used antibodies raised against whole parasite lysates (i.e., tachyzoites) have reported cross-reactivity in IHC for N. caninum and T. gondii.13,27 Recombinant proteins have provided an alternative method for producing more specific antibodies.15,36 We chose the TgSRS2 and NcSRS2 antigen genes for recombinant protein production because there is minimal amino acid sequence identity between the 2 regions chosen, making it more likely that they do not share B-cell epitopes and that antisera raised against these recombinant proteins would not cross-react with the other species. The SRS2 gene of N. caninum, and its homologue in the closely related T. gondii, encode proteins that have only 43% identity, which made these good candidates for the development of species-specific antisera.8,29 Our results are supported by findings of other studies, in which recombinant NcSRS2 surface antigens were not recognized by T. gondii immune cat, cattle, and mouse sera.6,18 It was demonstrated 18 that, even though TgSRS2 is a homologue of NcSRS2 and shares structural similarities, the levels of amino acid sequence identity of TgSRS2 and NcSRS2 were not sufficient to elicit a cross-reactive antibody response between the antigens. Our results indicate that the recombinant proteins generated did not have any cross-reactivity between bovine positive N. caninum and T. gondii sera and were therefore a good target for the development of polyclonal antibodies. However, it was surprising that negative control cattle sera reacted weakly with NAF or TAF. These sera originated from experimental studies conducted at the Moredun Research Institute and were thought to be uninfected with N. caninum and T. gondii. 35
The development of polyclonal sera was initiated by screening the rabbit pre-immune sera to choose the best candidate for immunization with recombinant proteins. The screening of pre-immune sera revealed that only 3 of 14 rabbits could be suitable for polyclonal serum production given that they did not react with N. caninum or T. gondii by either western blot or IHC analyses. The positive rabbits may have had antibodies against other biologic material that either recognized the parasite epitopes directly, or nonspecifically cross-reacted with parasite epitopes. Rabbits are known to produce large amounts of nonspecific antibodies, creating high background signals during IHC analysis. 23 Many environmental factors can encourage immune responses prior to immunization against various pathogens (i.e., bacteria, fungi, viruses). 36 An ELISA study 23 showed that 53% of the rabbit pre-immune sera had positive reactions with 10 or more bacterial cultures. These and our results suggest that the presence of cross-reactive antibodies in pre-immune sera against microorganisms such as bacteria and protozoa is an important common problem that should be considered when using rabbits to develop specific antisera. 23
Our results from the rabbit polyclonal antisera, tested for their specificity by western blot analyses, showed that the rabbit anti-rNcSRS2 serum only reacted with NAF and rNcSRS2. This indicates that the serum specifically recognizes N. caninum, making it a good candidate for IHC, given that no reactivity with rTgSRS2 or TAF was seen. The rabbit anti-rTgSRS2 serum reacted only with rTgSRS2, indicating that the polyclonal serum is specific to the T. gondii protein. However, the lack of reactivity with TAF suggests that the rTgSRS2 may not have displayed native epitopes, as a result of the refolding of the recombinant protein during dialysis and following immunization. Further tests could be conducted to see if these polyclonal antibodies would also make good candidates for other serologic assays, such as ELISAs or indirect immunofluorescence assays. A study has shown that it is important that proteins used are homogeneous, correctly folded, and presented so that the critical epitopes are accessible. 11 Each recombinant protein expressed in our study was insoluble and, even though refolding procedures were used, some proteins may have not been able to reproduce the properties of the native protein. 9 Protein oxidation, aggregation, and degradation are known issues that can severely affect the outcome of antibody generation, and therefore it was important to test functionality and specificity of the polyclonal sera using more than one assay (i.e., western blots, IHC). 11
We further evaluated each polyclonal serum using archived tissues from ruminants (sheep and cattle), and the results demonstrated that the polyclonal antiserum could be used for the specific detection of N. caninum and T. gondii in FFPE tissue from naturally occurring protozoan cases. The level of agreement between IHC and PCR results was high for both T. gondii and N. caninum (overall agreement 90.1%). In all cases, the agreement was statistically significant (kappa of 0.42–0.64), suggesting that both tests can be used for the detection of N. caninum and T. gondii. However, it is worth remembering that although PCR analysis is more sensitive, it can only detect genomic DNA whereas IHC can detect the parasite within the tissue samples, thus providing useful information on pathogen location and distribution and allowing visualization of the host tissue response. Findings that some samples were positive by one method and not the other may be the result of the impossibility of testing the exact same sections by PCR and IHC, hence giving variable results of cases being positive by one test but negative by the other and vice versa. In general, protozoan parasites are unevenly distributed in tissue samples, making it generally more difficult to obtain matching results unless multiple FFPE scrolls are used with each test. 1
The chances of labeling and detecting N. caninum or T. gondii using a single tissue block and single section can be low compared to using multiples and thicker sections, and the true number of positives of these protozoan parasites in ruminants could be underestimated. In our study, the percentage of negative results could be explained by only testing one random section from one block with each antiserum, resulting in false-negative results for the animal. It has been shown 1 that certain areas of the ovine brain had a higher density of T. gondii parasites than others, and that protozoan tissue cysts and antigen were recorded more frequently in the forebrain and midbrain. Hence, for diagnostic purposes, consideration should be given to the use of multiple sections per block and several blocks per animal. Moreover, blocks in which the protozoan parasites are most likely to occur should be tested more frequently to decrease false-negative results. The limited number of positive IHC results compared to PCR results can be explained by the size of the sample for IHC, which was a 4-µm section from a single block (and not always from the same tissue block) compared to 20 µm for PCR, decreasing the chance of parasite detection considerably given that parasite distribution is not homogeneous. 27 Moreover, tissue morphology changes considerably with each section cut, and parasites seen in one section might be absent in the following section, especially as these protozoan parasites can be smaller than 4 µm.
Supplemental Material
sj-pdf-1-vdi-10.1177_10406387241234322 – Supplemental material for Evaluation of species-specific polyclonal antibodies to detect and differentiate between Neospora caninum and Toxoplasma gondii
Supplemental material, sj-pdf-1-vdi-10.1177_10406387241234322 for Evaluation of species-specific polyclonal antibodies to detect and differentiate between Neospora caninum and Toxoplasma gondii by Tanja Lepore, Alastair I. Macrae, Germán J. Cantón, Carlo Cantile, Henny M. Martineau, Javier Palarea-Albaladejo, Stephen Cahalan, Clare Underwood, Frank Katzer and Francesca Chianini in Journal of Veterinary Diagnostic Investigation
Footnotes
Acknowledgements
We thank Val Forbes for excellent technical support.
Declaration of conflicting interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
Our study was funded by the Biotechnology and Biological Science Research Council (BBSRC), Zoetis, and the Scottish Government’s Rural and Environment Science and Analytical Services Division (RESAS). For open access, the authors have applied a Creative Commons Attribution CC-BY license to any Author Accepted Manuscript version arising from this submission.
Supplemental material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.