
Abstract
Almost all mitochondrial proteins are encoded in the nuclear DNA and synthesized in the cytosol as pre-proteins. There is a protein translocase located in the mitochondrial outer membrane that transports mitochondrial pre-proteins into mitochondria. The central component of this translocase of the outer mitochondrial membrane (TOMM) complex is TOMM40, and TOMM5 is one of three small subunits associated with TOMM40. Translocase of outer mitochondrial membrane 5 homolog (Tomm5–/–) knockout mice demonstrated an unexpected lung-specific phenotype characterized by widespread intra-alveolar fibrosis. Although TOMM5-deficient mice tested normal in a very broad range of phenotyping assays, they displayed histopathological lesions in the lung that were consistent with those reported in humans with cryptogenic organizing pneumonia (COP), which is also known as bronchiolitis obliterans organizing pneumonia (BOOP). The lesions had a patchy distribution in the lung and were characterized by the presence of intraluminal fibrogenic buds consisting of fibroblasts and myofibroblasts embedded in a loose connective tissue matrix that occupied the lumina of alveoli and alveolar ducts, with preservation of underlying alveolar architecture. In addition to macrophages, which were numerous in affected and surrounding alveoli, eosinophils comprised the most common and widespread inflammatory cell. Taken together, the findings in Tomm5–/– mice provide yet another example of the value of histopathology as a baseline assay in high-throughput phenotyping systems.
Keywords
Mitochondria contain between 1000 and 2000 different proteins that are encoded by either nuclear or mitochondrial DNA. Given the very small number of proteins encoded by mitochondrial genes, it becomes obvious that the vast majority of mitochondrial proteins are encoded in the nuclear DNA. Since these nuclear-encoded mitochondrial proteins are synthesized in the cytosol as preproteins, a mechanism must exist to transport them into mitochondria. This transport is accomplished via a protein translocase located in the mitochondrial outer membrane. This translocase of the outer mitochondrial membrane (TOMM) complex has multiple functions; it recognizes the precursor proteins in the cytosol, facilitates the release of their cytosolic binding factors, contributes to the unfolding of cytosolic protein domains, and transfers the polypeptides across the outer mitochondrial membrane. The TOMM complex consists of a translocation pore, which consists of membrane-embedded subunits (TOMM40, TOMM7, TOMM6, and TOMM5), plus several loosely attached receptor subunits (TOMM70, TOMM22, and TOMM20)19,29,34 that pass precursor proteins to the translocation pore. 44 The central component of the TOMM complex is TOMM40, which forms the β-barrel protein translocation channel. 42 Even in the absence of other TOMM subunits, purified TOMM40 is able to form membrane pores that show similar functional characteristics to those of the entire TOMM complex. 8 The precise functions of the three small subunits (TOMM5, TOMM6, and TOMM7) associated with TOMM40 are not fully understood, but they appear to be involved in maintaining the stability of the complex. 42 The function of TOMM5 varies slightly in different species. In Saccharomyces cerevisiae, assembly of TOMM40 from precursors depends on TOMM5. 9 TOMM5 also has a receptor function in the transfer of precursor proteins from TOMM22 to the TOMM40 pore, as depletion of TOMM5 inhibits the mitochondrial import of precursor proteins. 20 In contrast, in Neurospora crassa the stability of the TOMM complex and protein import does not appear to be affected by the depletion of TOMM5. 50 In human cells, both TOMM5 and TOMM6 have only recently been localized to the TOMM complex in the mitochondrial outer membrane, where they appear to help maintain the structural integrity of the TOMM complex. 32 TOMM5 also plays a role in the final assembly and placement of the mature β-barrel forming protein TOMM40 in the mitochondrial membrane via its association with the TOMM40 precursor at an early stage of the assembly process. 9
In order to identify novel genes that code for pharmaceutically relevant disease targets, Lexicon Pharmaceuticals initiated its Genome5000™ program to produce and phenotype knockout mice.56,58 During the high-throughput mutagenesis and phenotyping of over 4650 knockout mouse lines, we discovered many phenotypes in mice that have proven useful in elucidating fundamental processes in biology and the pathogenesis of disease. We report here some notable pathologic findings in TOMM5-deficient (Tomm5 Gt(OST44663)Lex abbreviated as Tomm5–/–) mice. Although TOMM5-deficient mice were essentially normal according to all other phenotyping assays, we found that they displayed histopathological lesions in the lung that were consistent with those reported in humans with cryptogenic organizing pneumonia (COP), which is also known as bronchiolitis obliterans organizing pneumonia (BOOP). These important findings in otherwise apparently normal mice provide yet another example of the value of including histopathology analysis as a baseline assay in high-throughput phenotyping systems. 48 Although we did not investigate the molecular mechanisms involved in the pathogenesis of COP in TOMM5-deficient mice, our findings indicate that this animal model could be very useful in research studies intended to elucidate the processes involved in the pathogenesis of pulmonary lesions in COP.
Materials and Methods
Mouse Production
Methods for gene trapping in embryonic stem (ES) cells, identification of trapped genes using Omnibank® Sequence Tags (OST), characterization of retroviral gene-trap vector insertion site, and Reverse transcription polymerase chain reaction (RT-PCR) analysis of knockout and wild type transcripts are published.56,57 This gene trap allele of Tomm5 was generated in 129S5SvEvBrd-derived ES cells obtained from the OmniBank library as described previously. 56 OST44663 was selected for mutation analysis based on sequence similarity between the 3′ RACE tag (CG501096) obtained from this clone and the Tomm5 gene (accession NM_ 001099675). An intragenic gene trap mutation in this clone was confirmed by identifying the precise location of vector insertion using inverse genomic PCR. 1 Briefly, oligonucleotide primers complementary to the gene trap vector were used to amplify the vector insertion site from this clone, which was then compared to mouse genome sequence assemblies to localize the insertion with respect to the exons and introns of the gene. Gene disruption in vivo was confirmed by a direct analysis of gene expression using RT-PCR. For this, RNA was extracted from kidney and spleen of wild-type and mutant mice using a bead homogenizer and RNAzol (Ambion, Austin, TX) according to manufacturer’s instructions. Reverse transcription was performed with SuperScript II (Invitrogen, Carlsbad, CA) and random hexamer primers, according to the manufacturer’s instructions, with PCR amplification being performed with oligonucleotide primers complementary to exons flanking the insertion site (F: 5′- GGAGGAGATGAAGCGGAAGATGCGTGAGG-3′ and R: 5′-AGCTGCAGACAGGAGGAGAAGCCGTCACCA-3′). RT-PCR products were gel purified and verified by direct sequencing. Mutant mice were generated by microinjection of ES cells into C57BL/6-Tyr c-Brd (albino) blastocysts to generate chimeric animals that were bred to C57BL/6-Tyr c-Brd (albino) females, and the resulting heterozygous offspring were interbred to produce homozygous gene-deficient mice. Knockout F2 mice used in phenotyping studies were produced by intercrossing the F1 heterozygous knockout (–/+) offspring of chimeric founder parents. Therefore, all mice used in these studies are of mixed C57BL and 129 genetic backgrounds. In all studies reported here, mutant mice were compared directly with their wild-type littermates used as negative controls. Using the albino variant of C57BL/6 mice (C57BL/6-Tyr c-Brd ) permits simple visual recognition of chimeric offspring, because they have dark eyes and patches of dark hair that derive from stem cells from the agouti 129S5/SvEvBrd. Genotypes of offspring were determined as previously described 28 by quantitative PCR of DNA isolated from tail biopsy samples for the neo gene, which is present in the gene trapping vector. Genotypes were subsequently confirmed with a mutation-specific PCR assay using oligonucleotide primers (LTR-2: 5′- AAATGGCGTTACTTAAGCTAGCTTGC-3′ and OST44663 Rev: 5′-AGCCAGCAATACAGAGAGTACAGC-3′), which produce a mutant product of 220 bps. Inverse genomic PCR was used to amplify the vector insertion site from this clone and localize the insertion mutation to intron 1 of the gene (Fig. 1a). The insertion was determined to occur downstream of the single initiation codon of Tomm5 (Fig. 1a), suggesting that both isoforms were disrupted.
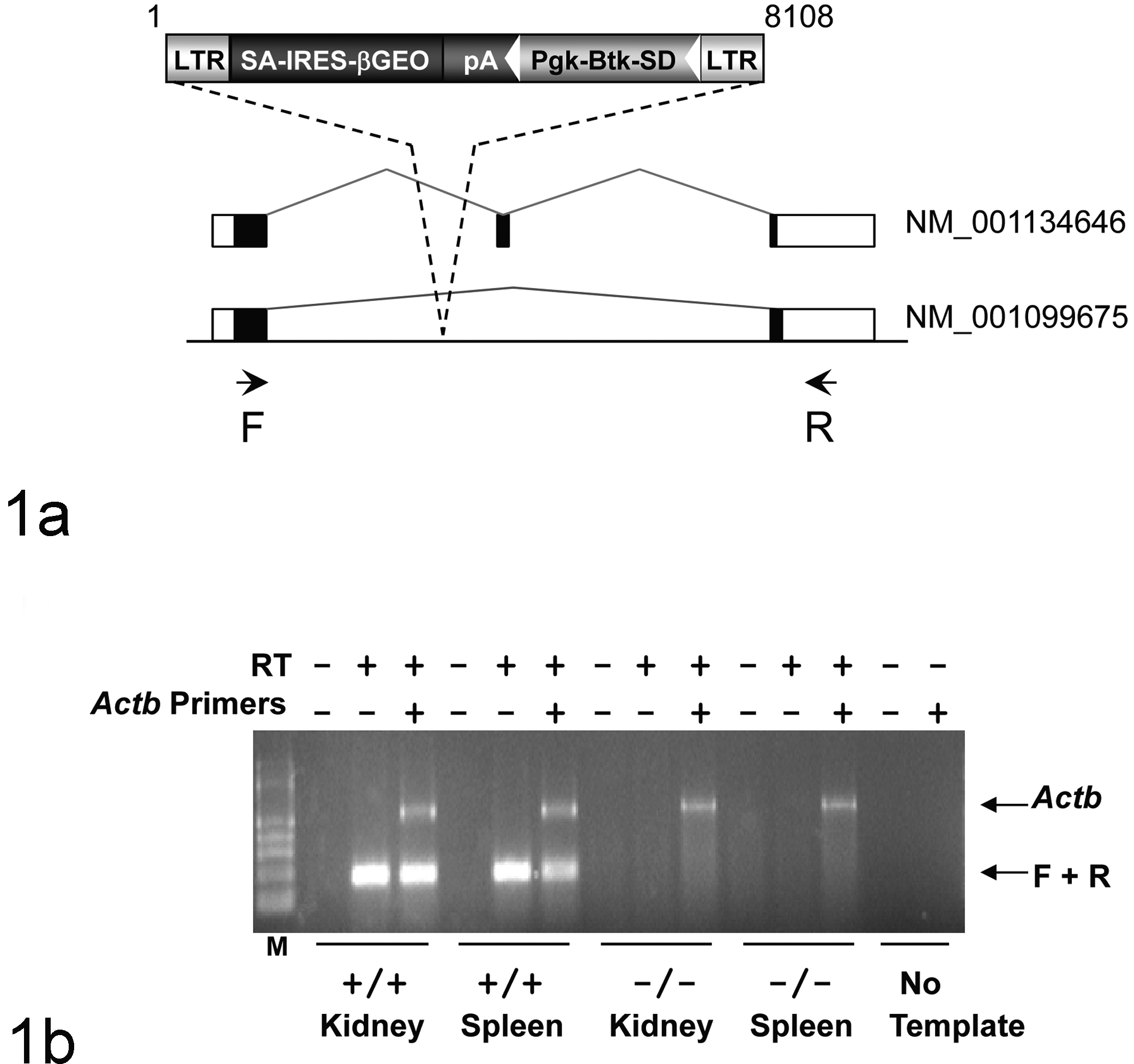
Generation of Tomm5Gt(OST44663)Lex mice. (a) Retroviral gene trap vector VICTR37 was used to produce OmniBank clone OST44663 (accession CG501096), which contains an insertion within intron 1 of the Tomm5 gene. This mutation would be expected to truncate the Tomm5 gene product following the first coding exon, interrupting both transcript variants of the Tomm5 gene. Open boxes denote untranslated exons, filled boxed denote coding exons. LTR, viral long terminal repeat; SA, splice acceptor sequence; IRES, internal ribosomal entry site; βGEO, fusion of the beta-galactosidase and the neomycin phosphotransferase genes; pA, polyadenylation sequence; Pgk, phosphoglycerate kinase-1 promoter; Btk-SD, Bruton’s tyrosine kinase splice donor sequence. (b) RT-PCR expression analysis of Tomm5 transcript. Endogenous Tomm5 transcript was detected in the kidney and spleen of wild type (+/+) mice. No endogenous Tomm5 transcript was detected in homozygous (–/–) tissues. Primers F and R are complementary to Tomm5 exons 1 and 3, respectively, and amplify a product of 185 nucleotides (variant 1). RT-PCR analysis using primers (Actb) complementary to the mouse beta actin gene (accession number M12481) was performed in the same reaction as an internal amplification control. M, molecular weight marker.
Mouse Husbandry
Mice were housed in micro-isolator cages within a barrier facility at 24ºC on a fixed 12-hour light and 12-hour dark cycle and were provided ad libitum acidified water and Purina rodent chow No. 5001 (Purina, St. Louis, MO). Procedures involving animals were conducted in conformance with Lexicon’s Institutional Animal Care and Use Committee guidelines that are in compliance with the state and federal laws and the standards outlined in the Guide for the Care and Use of Laboratory Animals (National Research Council, 1996). Quarterly sentinel surveillance showed no evidence of pathogenic rodent viruses, Mycoplasma or Helicobacter spp. in the Lexicon Pharmaceutical source colonies.
Phenotypic Screening
Wild-type and homozygous null mice were subjected to a comprehensive battery of phenotype screening exams as previously described.11,56 Screening assays included behavioral tests (eg, circadian rhythm, open field, inverted screen, prepulse inhibition of the acoustic startle response, tail suspension, marble burying, and context trace conditioning), fundoscopy and retinal angiography exams, blood pressure and heart rate measurements, serum chemistries, insulin levels, glucose tolerance testing, urinalysis, Quantitative Magnetic Resonance, Dual-energy x-ray absorptiometry (DEXA) scans, computerized axial tomography (CAT) scans, microcomputed tomography (Micro-CT) scans, fertility testing, skin fibroblast proliferation assays, and pathology. Immunology assays included hematology, peripheral blood Fluorescence-activated cell sorting (FACS) analysis, acute phase response, and ovalbumin challenge.
Histopathology
Seven-week-old knockout (2 male and 2 female) and age-matched normal control mice (2 male and 2 female) were euthanized and fixed by cardiac perfusion with 10% neutral buffered formalin. Lungs were infused with formalin via the trachea, and all tissues were immersed 10% neutral buffered formalin for an additional 48 hours, except for the eyes, which were removed and fixed by immersion in Davidson’s fixative (Poly Scientific, NY) overnight at room temperature. All tissues were embedded in paraffin, sectioned at 4 µm, mounted on positively charged glass slides (Superfrost Plus, Fisher Scientific, Pittsburgh, PA), and stained with hematoxylin and eosin (HE) for histopathological examination. Masson’s Trichrome and Sirius red staining were used to visualize collagen deposition.
Immunohistochemistry
Immunohistochemistry was also performed on 4-µm-thick sections of formalin-fixed, paraffin-embedded tissues. The primary antibodies used in this study (and working dilutions) included: an anti-α Smooth Muscle Actin at 1:20 dilution (No. M0851, DAKO, Carpinteria, CA, USA) for myofibroblasts; an anti-fibronectin at 1:200 (No. sc6952, Santa Cruz Biotechnology, Santa Cruz, CA) for fibronectin; an anti-podoplanin GP38 antibody at 1:1000 dilution (No.14-5381, eBioscience, San Diego, CA) for type I pneumocytes; an anti-MECA32 antibody at 1:50 dilution (No. 553849, BD Biosciences, San Jose, CA) and an anti-CD34 antibody at 1:50 dilution (No. 553731, BD Biosciences, San Jose, CA) for endothelium; an anti F4/80 at 1:1000 dilution (No. MS48000, Caltag, Burlingame, CA) and an anti-MAC2 antibody diluted 1:20 000 (No. ACL-8942AP, Accurate, Westbury, NY) for macrophages; an anti-MBP (anti-major basic protein, kindly provided by Mayo Clinic, Rochester, MN) for eosinophils.
Results
Phenotype Screening Findings
In the production colony, the increased mortality observed in pups was shown to be largely due to decreased survival of neonatal Tomm5-deficient mice. At the time of genotyping (3 weeks of age), the number of surviving knockout mice (77 wild type: 164 heterozygote: 53 homozygote) was reduced below expected Mendelian ratios. There were a few additional unexpected deaths of Tomm5-deficient mice between 4 and 7 weeks of age, but additional deaths or clinically ill mice were not observed after phenotyping assays began at 8 weeks of age. After this time, Tomm5-deficient mice were indistinguishable from wild-type littermates upon macroscopic examination and showed normal growth rates and fertility. In addition, they showed normal results in behavioral tests (eg, circadian rhythm, open field, inverted screen, prepulse inhibition of the acoustic startle response, tail suspension, marble burying, and context trace averse conditioning), fundoscopy and retinal angiography exams, blood pressure and heart rate measurements, serum chemistries, insulin levels, glucose tolerance testing, urinalysis, Quantitative Magnetic Resonance, DEXA scans, CAT scans, Micro-CT scans, and skin fibroblast proliferation assays. Immunological assays included hematology, peripheral blood FACS analysis, acute phase response, and ovalbumin challenge, and standard serum chemistry and hematology workups were unremarkable. The only abnormal findings detected in phenotype screening were in histopathology, where lesions were found in the lungs and thymus of 4 mice submitted for necropsy at 6 to 7 weeks of age due to clinical signs of lethargy. To confirm the disruption of Tomm5 transcription by the gene trapping vector, we performed RT-PCR using primers complementary to exonic sequences flanking the vector insertion site. No endogenous Tomm5 transcript was detected in Tomm5–/– tissues (Fig. 1b).
Histopathological Findings
The key histological finding in Tomm5–/– mice was the presence of patchy intra-alveolar organizing pneumonia (Fig. 2). Interestingly, these pulmonary lesions were much more common in the four lung lobes on the right side (the cranial, middle, caudal, and accessory lobe) than in the left lobe, where lesions were relatively rare and when present, were generally restricted to peripheral alveolar ducts and subpleural alveoli.
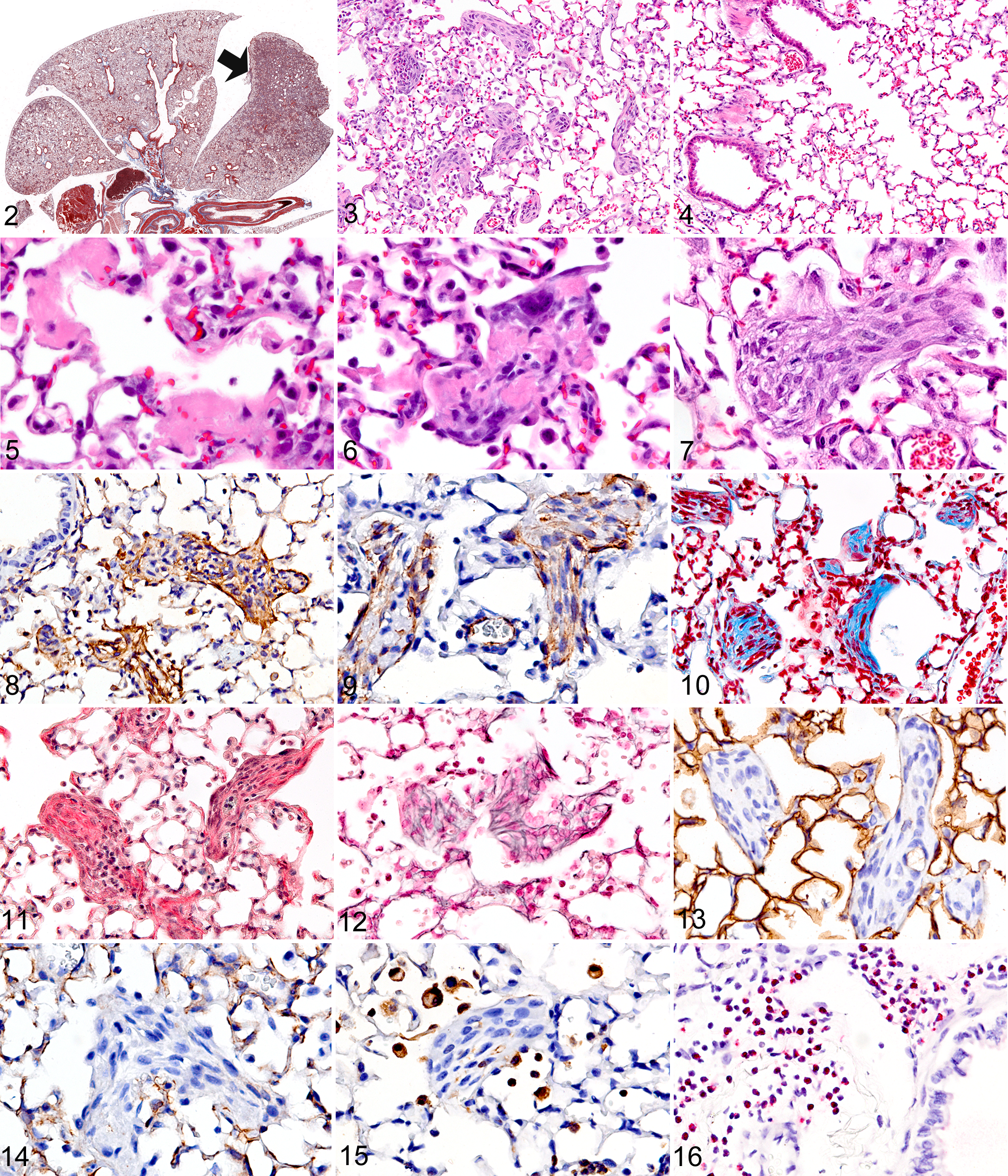
In most affected areas, pulmonary lesions consisted of clearly defined and separated foci of intra-alveolar fibrogenesis (Fig. 3). In areas adjacent to the affected lung tissue and in most of the left lobe, the alveoli appeared to be essentially normal (Fig. 4). The pulmonary lesions in TOMM5-deficient mice could be categorized into three types of proliferative intra-alveolar fibrosis. In the first form, the intra-alveolar pulmonary fibrosis was characterized by well-defined intraluminal projections consisting of collagen-embedded myofibroblasts/fibroblasts that were attached to alveolar walls as thin-stalked polypoid buds. In the second form, sessile intra-alveolar fibroblastic buds were broadly attached to thickened alveolar septa. The third type of intra-alveolar fibrosis was characterized by focally extensive areas of obliterative change, where masses of myxoid fibrogenic tissue filled and obstructed the air spaces.
In the human medical literature, virtually identical intraluminal and mural fibrogenic buds or plugs in distal airways, characterized by whorls of myofibroblasts in a connective tissue matrix, were identified many years ago 41 and are called “Masson bodies.” In Tomm5–/– mice, Masson bodies were common in the areas of lungs affected by either of the first two forms of intra-alveolar fibrosis, which are both characterized by multifocal intraluminal buds. In these areas, the fibrogenic tissue sometimes extended from one alveolus to the next through the narrow pores of Kohn, to form bridges between adjacent alveoli and alveolar ducts (butterfly pattern). Although inflammatory cells were generally absent in Masson bodies, some contained small numbers of mixed inflammatory cells, while clusters of eosinophils were predominant in a few. Numerous alveolar macrophages were always present within the alveoli surrounding affected areas, but variable numbers of eosinophils were often present in the interstitium and airways. Interestingly, eosinophils tended to be most numerous in pulmonary tissues located more distant from the fibrosing lesions, such as the peribronchiolar connective tissues in the left lobe of the lung. Notably, despite sometimes extensive intra-alveolar fibrosis, there was little disruption and virtually no destruction of the normal-appearing preexisting lung architecture. Neovascularization was conspicuously absent in the areas of organizing pneumonia; capillaries were not seen within the typically avascular cores of Masson bodies but were often located along their base within preexisting septal stroma. Although fibrogenic lesions were widespread in alveoli and alveolar ducts, similar lesions were not present in bronchioles, and bronchiolitis obliterans was not a feature seen in the lungs of Tomm5–/– mice.
The less common third form of intra-alveolar fibrosis in these mice was characterized by extensive consolidated areas of lung where the lumens of partially collapsed alveoli were filled with newly formed fibromyxoid tissue. These consolidated areas of pulmonary fibrosis could be extensive, but even in areas of diffuse intra-alveolar fibrosis there was preservation of the preexisting pulmonary architecture (alveolar septa) and generally only minimal amounts of interstitial fibrosis and inflammation, with alveolar macrophages again being the predominant inflammatory cell component but eosinophils also being present.
In order to better characterize these lesions, a variety of histochemical stains and immunohistochemical assays were completed. Trichrome and reticulin stains effectively highlighted the extent of intra-alveolar collagen and reticulin formation and were very useful in elucidating the progression of lesion development and the maturation of Masson bodies. Although most of the lesions within a given lobe appeared to be at the same stage of development, there were clear differences in lesions between animals and even between individual lobes, providing an opportunity to evaluate the temporal progression of intra-alveolar lesions.
At the earliest stage, lesions were characterized by multifocal deposits of fibrin, which was often arranged in thick layers covering alveolar surfaces (Fig. 5). The fibrin deposits were initially acellular, but in some foci myofibroblasts could be seen infiltrating and organizing the fibrinous exudates into cellular intraluminal buds and masses (Fig. 6). The more acute and consequently less-organized intra-alveolar lesions contained abundant fibronectin (Fig. 7), which was gradually replaced over time by cellular aggregates of fibroblasts/myofibroblasts (Fig. 8) and collagen. Inflammatory cells also became progressively less common in developing fibrogenic bodies, although small aggregates of inflammatory cells could always be found in some, presumably due to entrapment. Immunohistochemical staining confirmed that the early lesions contained abundant fibronectin (Fig. 9) and that most of the cells in the less mature but highly cellular fibrogenic buds/plugs were α-SMA positive myofibroblasts (Fig. 10). At early stages, the expanding fibrogenic bodies were still largely negative for collagen and reticulin (as shown by Trichrome and reticulin stains, respectively). In contrast, the core of mature relatively hypocellular Masson bodies contained abundant collagen deposits as shown by Trichrome (Fig. 10) and Sirius Red histochemical stains (Fig. 11) as well as reticulin fibers by the Reticulin stain (Fig. 12). Mature buds were almost entirely negative for α-SMA (indicating the loss or differentiation of myofibroblasts), except for some lesions where much of the surface of intraluminal fibrogenic bodies was lined by α-SMA positive myofibroblasts. Conversely, immunohistochemical staining of type I pneumocytes with anti-podoplanin antibodies showed that fibrogenic bodies were at most only partially covered by type I pneumocytes (Fig. 13). Because capillarization of fibrogenic lesions is supposed to help differentiate cryptogenic from secondary organizing pneumonia, we stained sections with the endothelial cell markers CD34 and MECA32. These stains confirmed our earlier observation that capillaries were almost entirely absent within the core of intra-alveolar fibrogenic bodies (Fig. 14).
In the areas of fibromyxoid consolidation and obliterative change, intra-alveolar fibronectin and α-SMA positive myofibroblasts were abundant, but collagen and reticulin deposition was minimal, suggesting that these lesions represent a more acute or more slowly organizing process. Eosinophils also tended to be less common in these areas of fibromyxoid consolidation.
Although interstitial inflammation was minimal to mild in most areas of lung affected by organizing pneumonia, alveolar macrophages (F4/80 and MAC2 positive) were always abundant within affected alveoli and in surrounding air spaces (Fig. 15) but less common within the intra-alveolar fibrogenic lesions. Immunohistochemical staining for eosinophils (anti-MBP) confirmed that most of the granulocytes located in variable numbers throughout the lung were eosinophils. The distribution of eosinophils was uneven and patchy, with the most severe eosinophilic infiltrates being located within the peribronchial connective tissues and at the periphery of lung lobes within subpleural alveoli and interstitium (Fig. 16). These eosinophilic infiltrates were often not collocated with areas of intra-alveolar fibrosis. In fact, some of the most severe perivascular infiltrates were located in the relatively spared left lung lobe. Although some fibrocellular buds contained clusters of eosinophils, most others in the same general area contained none. The only other notable histopathological findings in these clinically sick mice were thymic atrophy and bone marrow granulocytosis. The thymic atrophy was characterized by almost complete depletion of thymocytes in the thymic cortex and was interpreted as a nonspecific response to stress in clinically ill mice, while the diffuse moderate eosinophilic granulocytosis in the bone marrow was most likely related to the pulmonary disease.
Discussion
Although all cells and tissues in Tomm5–/– mice are deficient for this small protein, demonstrable pathological lesions were restricted to the alveoli, suggesting that TOMM5 activity is especially critical for mitochondrial function in the unique environment of the lung. We do not know why TOMM5-deficient alveolar epithelium and/or endothelium would be more susceptible to damage than any other tissue, but the presence of intra-alveolar fibrosis in lungs of these mice indicates that cells comprising the alveolar septum have been damaged. It is also possible that the excessive fibrogenic response to injury could be due to aberrant signaling or improper control of fibrosis, favoring persistent injury and enhanced profibrotic influences instead of normal repair processes. In other types of pulmonary fibrosis, alveolar injury is generally initiated by inhalation of antigens or noxious materials or ionizing radiation. In contrast, the widespread alveolar damage in lungs of Tomm5–/– was apparently caused by exposure to agents or environmental factors that are innocuous in wild type mice. The consistent involvement of the right lung lobes and peripheral alveoli, combined with the relative sparing of the left lobe, suggests that either localized differences in the left side versus the right side lung environments, such as asymmetric and regional differences in the effectiveness of pulmonary clearance, perfusion or ventilation, or unique pneumocyte metabolic demands, might be responsible for the increased susceptibility of alveoli in these areas.
The association of TOMM5 deficiency with an organ-specific disease process was unexpected. Relatively few diseases have been linked to defective mitochondrial protein transport, 39 probably because functional mitochondria are essential to cell survival and interference with the transport process would usually result in mitochondrial dysfunction and cell death. Given the central role of mitochondria in cell survival, it is not surprising that those heritable diseases identified thus far that involve mitochondrial protein transport deficiencies have all been multisystemic syndromes; these include dilated cardiomyopathy with ataxia, human deafness dystonia syndrome, and spastic paraplegia. 39 On the other hand, it is likely that effects of acquired defects in mitochondrial protein import that are reported to occur with aging, oxidative stress, and thyroid hormone (T3) treatment are also important (reviewed in MacKenzie and Payne 39 ). Nonetheless, until now no disease process has been linked to defective TOMM5, but our findings in this mouse model raise the possibility that defective alleles of Tomm5 could contribute to increased susceptibility to some pulmonary diseases in humans and other animals.
We did not determine how TOMM5 deficiency leads to alveolar damage in mice, but speculate that functional TOMM5 may be particularly important for normal mitochondrial protein transport in alveolar epithelium during the early neonatal adjustment period to a relatively high oxygen environment. Air breathing exposes the lung to reactive oxygen species, and hyperoxia has been shown to increase oxygen radical production in rat lungs and lung mitochondria. 24 Deleterious effects of oxidative stress on mitochondrial preprotein transport have been shown to be due to inhibited import of precursor proteins and their subsequent degradation. 55 Oxidative stress has also been shown to decrease the mitochondrial membrane potential and membrane fluidity and to alter the expression of genes involved in mitochondrial bioenergetics. 51 Clearly, reduced mitochondrial function could delay repair processes, allowing injury to persist. Another possibility is that TOMM5 deficiency favors the development of a hypersensitivity to some environmental agent commonly present in a mouse cage (eg, particulates, ammonia).
Given the extent of pulmonary lesions found in the 4 clinically ill mice described in this report, it is likely that the decreased viability of neonatal Tomm5–/– mice was due to acute respiratory distress. Unfortunately, no Tomm5–/– pups were submitted for histopathological evaluation to evaluate this possibility. However, in humans a rapidly progressive cryptogenic organizing pneumonia can be fatal within 1 to 3 days of the onset of signs and symptoms of acute onset of acute respiratory distress syndrome.21,23 Evaluation of Tomm5–/– mice under controlled conditions in the neonatal period might help identify the precipitating cause of alveolar damage.
The development of intra-alveolar fibrosis in Tomm5–/– mice provides a unique animal model of pulmonary fibrosis that should be useful for advancing our understanding of the pathogenetic mechanisms involved in the development of pulmonary fibrosis. In contrast to the apparently spontaneous development of pulmonary fibrosis in these knockout mice, virtually all other mouse models of pulmonary fibrosis require the direct administration of a profibrotic agent to the lungs. The wide range of exogenously administered agents that can induce pulmonary fibrosis in mice includes bleomycin, irradiation, fluorescein isothiocyanate, vanadium pentoxide, haptenic antigens, and inorganic particles (silica, asbestos) and infectious agents. 13 While all of these agents generate varying degrees of parenchymal lung scarring, none of them induce the progressive forms of pulmonary fibrosis that are of greatest concern in humans. 13 Nevertheless, much of recent research into the cellular interactions and molecular pathways involved in lung tissue repair and fibrosis, as well as the development of novel therapeutic approaches to treating pulmonary fibrosis, has relied on interpreting findings from these mouse models. 13
In humans, fibrosing pulmonary disorders can follow exposure to a wide range of insults, including radiation and chemotherapy, infections, immunologic reactions, gene mutations, smoking, dust, and exposure to other toxic and physical insults (reviewed in Schwartz and King 49 ). However, the underlying cause of pulmonary fibrosis is unknown in many cases, which are then diagnosed as idiopathic interstitial pneumonias. The idiopathic interstitial pneumonias have been classified into seven entities. These are idiopathic pulmonary fibrosis, nonspecific interstitial pneumonia, acute interstitial pneumonia, respiratory bronchiolitis-associated interstitial lung disease, desquamative interstitial pneumonia, lymphoid interstitial pneumonia, and cryptogenic organizing pneumonia. 3 The predominant or only type of pathological pulmonary fibrosis in most of these entities is interstitial fibrosis associated with interstitial pneumonia or diffuse alveolar damage, the lone exception being cryptogenic organizing pneumonia, which is instead characterized by intra-alveolar fibrosis. Despite the fact that the pulmonary lesions in COP are mainly intra-alveolar, COP is classified with the other idiopathic interstitial pneumonias because of: (1) its idiopathic nature (cryptogenic is synonymous with idiopathic), (2) its clinical and radiological similarities to the other forms of idiopathic interstitial pneumonias, and (3) the presence of interstitial inflammation in the involved areas. 3 It should be noted that the current classification system for interstitial pneumonias recommends the term COP instead of BOOP, because COP more accurately describes the major features of the syndrome and also avoids confusion surrounding “bronchiolitis obliterans” in the term BOOP, since this specific lesion is often minimal or absent. 3
The intra-alveolar fibrosis of the organizing pneumonias represents a unique type of inflammatory lung disease. Organizing pneumonias can develop in association with connective tissue diseases, a variety of drugs, malignancy, ionizing radiation, and other interstitial pneumonias6,22 or be idiopathic.18,23 The intra-alveolar buds in organizing pneumonia share some morphological features with the newly formed connective tissue that develops in distal air spaces in other forms of interstitial pneumonia, 33 but while most other fibrosing pulmonary diseases are associated with progressive irreversible fibrosis with no effective treatment and a fatal outcome, 3 the prognosis for cryptogenic organizing pneumonia is good, with most patients recovering rapidly with corticosteroid therapy. 15
Of the previously listed interstitial pneumonias, the clinical history and histopathological findings we observed in Tomm5–/– mice are most similar to those reported in human COP. Cryptogenic organizing pneumonia was first described as a distinct clinicopathological entity of unknown origin in 1983, 18 with the same condition being named bronchiolitis obliterans with organizing pneumonia by Epler et al. 23
The key histopathological features that are used to distinguish COP from other pulmonary diseases in humans were also observed in Tomm5–/– mice. COP in humans is characterized histopathologically by the presence of patchy distribution of fibroblastic tissue proliferation consisting of fibroblasts and myofibroblasts embedded in a loose connective matrix that occupies the lumina of the distal airspaces, including alveoli, alveolar ducts, and bronchioles.14,18,23,38 These lesions generally present a uniform, temporally recent appearance, with no severe disruption of the preexisting lung architecture. Severe interstitial fibrosis, giant cells, granulomas, and vasculitis are rare or absent. In humans, the accumulation of foamy macrophages in the alveolar spaces have been attributed to bronchiolar occlusions, 49 which are not a feature of the disease in mice. Nevertheless, in Tomm5–/– mice we also observed a patchy distribution, the presence of intraluminal buds of proliferative fibrogenic tissue in the distal airspaces (alveoli and alveolar ducts, but unlike in human, bronchioles were unaffected) with preservation of normal lung architecture, abundant alveolar macrophages in affected areas, and minimal to mild interstitial inflammation. Similarly, the three different forms of proliferative intra-alveolar fibrosis that we observed in Tomm5–/– mice were virtually identical to three different patterns of intra-alveolar fibrosis originally reported in histopathological studies of COP in humans; these included: (1) intraluminal buds, which partially filled the alveoli, alveolar ducts, and/or distal bronchioles; (2) mural incorporation of intraluminal connective tissue masses, which have fused with alveolar and/or bronchiolar structures, and (3) obliterative changes, in which proliferative connective tissue masses fill the lumens of alveoli, alveolar ducts, or distal bronchioles. 7
Although all 4 Tomm5–/– mice evaluated in this report were all approximately the same age (7 weeks), and the fibrotic lesions within a given lobe appeared to be at the same stage of development, the presence of clear differences in lesion maturity between animals and even between individual lobes permitted us to track the temporal progression of lesion development. The stages of lesion development we observed matched what is known about the pathogenesis of COP that has been learned from previous studies in other experimental animal models.15,25 These models include a mouse model that develops follicular bronchiolitis and intra-alveolar fibrosis following intranasal inoculation of relatively low doses of Reovirus serotype 1 into CBA/J mice. Notably, mice exposed to a 10-fold higher dose of the same virus develop histological lesions of acute respiratory distress syndrome, characterized by diffuse alveolar damage, hyaline membranes, and intra-alveolar fibrosis. Apparently a milder infection of pulmonary epithelium is a critical requirement for the development of COP. 10 T cells are also necessary in this Reovirus-infected mouse model for the development of intraluminal fibrosis associated with COP. 40 A rat model utilizing a single intratracheal exposure to bleomycin also produces lesions of intra-alveolar fibrosis. 53 Studies in both of these animal models have shown that the process of intra-alveolar organization follows a sequence initiated by damage to alveolar septal epithelium and capillary endothelium, followed by the leakage of plasma proteins, including coagulation factors, into the alveolar lumen. The same temporal evolution of intra-alveolar fibrosis has been described in human organizing pneumonia.35,43,46
Alveolar epithelial injury is the first event, with necrosis and sloughing of pneumocytes resulting in the denudation of the epithelial basal laminae. Most basal laminae are not destroyed, although some gaps are present and endothelial cells are only mildly damaged. Leakage of plasma proteins (including clotting factors) into the airspaces is crucial to the development of COP. 15 At the earliest stages, we observed multifocal lesions characterized by multifocal deposits of fibrin covering alveolar surfaces. It has been demonstrated that increased procoagulant activity and concurrent decreased fibrinolytic activity in damaged alveoli results in the deposition and persistence of fibrin in airways, 27 which provides a suitable matrix for organization by activated myofibroblasts that attach to the luminal side of epithelial basement membrane and produce intra-alveolar fibrosis and coalescence of alveolar walls. 25
Following the primary injury to the alveolar epithelium and basement membrane, and the subsequent deposition of fibrin, there is an early proliferative stage in which activated fibroblasts and myofibroblasts migrate through gaps in the epithelial basement membrane into the air spaces.7,35 The origin of these activated myofibroblasts is not clearly established, with resident intrapulmonary fibroblasts, bone marrow–derived fibrocytes, and even alveolar epithelial cells proposed as potential sources.17,30,52,54 Regardless of their origin, activated myofibroblasts migrate into the airspace and proliferate in response to a number of chemotactic, activation, and mitogenic substances that are released in the damaged alveolus.4,26 We observed that the initially acellular intra-alveolar fibrin deposits were rapidly infiltrated and organized by α-SMA positive myofibroblasts into developing fibrogenic bodies. At this stage of development, the expanding hypercellular fibrogenic bodies were still largely negative for collagen and reticulin (as shown by Trichrome and Reticulin stains, respectively).
In the final stage of organization, “mature” buds became relatively hypocellular and contained a core of abundant collagen and reticulin fibers. By this time, small clusters of inflammatory cells persisted as entrapped cells in some fibrogenic lesions, but there was no longer any fibrin within the alveolar lumen. Our histopathological findings, which we confirmed with immunohistochemical stains for endothelium, indicated that the intra-alveolar fibrogenic lesions in Tomm5–/– mice were avascular at all stages of development. This finding is in accordance with those of Ranzani et al, who used CD34 for capillaries and α-smooth muscle actin for myofibroblasts to differentiate COP from secondary BOOP. 47 Interestingly, a separate group reported prominent capillarization 36 and wide expression of vascular endothelial growth factor and basic fibroblast growth factor in intra-alveolar buds in human COP. 37
In addition to macrophages, which were numerous in affected and surrounding alveoli, eosinophils comprised the most common and widespread inflammatory cells in the lungs of Tomm5–/– mice. Interestingly, they appeared to be most numerous in the peribronchial connective tissues in the relatively spared left lung lobe and at the margins of the patchy fibrosing lesions. They were not common in the alveolar septa, but their presence in the lungs of Tomm5–/– mice may be significant, because the mere existence of some bronchiolar or intra-alveolar buds is not sufficient to make a diagnosis of organizing pneumonia. The organization of intra-alveolar exudates is a nonspecific process that can occur in a variety of inflammatory lung diseases, 14 including in eosinophilic pneumonia and acute hypersensitivity pneumonitis. 45 In these diseases, the presence of clusters of eosinophils or occasional loosely formed granulomas, respectively, may be the only features distinguishing these conditions from pure COP. 45 Idiopathic acute eosinophilic pneumonia is a cause of acute respiratory failure2,5 characterized by numerous interstitial and lesser numbers of alveolar eosinophils, accompanied by hyaline membrane formation and interstitial widening, as well as type II pneumocyte hyperplasia, interstitial lymphocytosis, an organizing intra-alveolar fibrinous exudate, and perivascular and intramural inflammation without necrosis. Typical alveolar buds of granulation tissue are often present in chronic eosinophilic pneumonia, but unlike in COP they are not the dominant feature of pulmonary disease.12,16 In human COP, small numbers of eosinophils are not infrequently seen, but the presence of numerous eosinophils increases the possibility that either organizing chronic eosinophilic pneumonia or an organizing allergic reaction would be the correct diagnosis. However, since small lung biopsies are typically used to diagnose these diseases in humans, it is likely that an uneven distribution of eosinophils in lungs, similar to what we observed in mice, could account for the variable presence of eosinophils in human lung biopsies.
Taken together, our findings do not rule out a role for eosinophils in the pathogenesis of the organizing pneumonia in Tomm5–/– mice. Despite the presence of eosinophils in lungs and the diffuse moderate eosinophilic granulocytosis in the bone marrow, we favor a diagnosis of COP rather than acute eosinophilic pneumonia for the pulmonary disease that developed in Tomm5–/– mice because intra-alveolar fibrosis was clearly the dominant histopathological finding. Eosinophilia was not detected during phenotype screening of Tomm5–/– mice, but it is likely that the apparently healthy older knockout mice that were sampled had either recovered from or never developed pulmonary lesions. There are indications that COP in humans may represent part of a disease continuum that includes chronic eosinophilic pneumonia and hypersensitivity pneumonitis. Some human cases appear to be true hybrid cases between COP and chronic eosinophilic pneumonia or COP and hypersensitivity pneumonitis. 14 In addition, all three syndromes involve underlying immunological mechanisms as COP, and both acute and chronic eosinophilic pneumonias are uniformly responsive to corticosteroid therapy. 31 Additional studies on Tomm5–/– mice may prove very useful in exploring the pathogenetic relationships between these pathologically related pulmonary diseases.
Animal models have been immeasurably important in advancing our understanding of the pathogenetic mechanisms involved in pulmonary fibrosis. Particular advantages of this Tomm5–/– mouse model are that the disease develops spontaneously, inflammatory infiltrates are relatively mild and very similar to those in human COP, and most affected mice survive. These features will enable time course studies to be performed in this mouse model. Another general advantage of mouse models is that transgenic and other genetically engineered mouse lines are available to dissect the molecular mechanisms involved and identify the specific nature of inciting agents and pathogenetic mechanisms and cell types responsible for the development of COP. The Tomm5–/– mouse model should help elucidate the roles that inflammation, alveolar epithelial cells, and myofibroblasts play in the pathogenesis of COP and provide a ready means to test hypotheses. For example, the complete resolution of COP following treatment with corticosteroids has been well documented, 22 but the specific mechanisms by which corticosteroid induces the disappearance of fibrous intra-alveolar buds are not known and might have broader applicability to other fibrosing lung diseases. The model might also be useful to definitively determine the origin of the myofibroblasts involved in COP, whether from resident interstitial fibroblasts, bone marrow fibrocytes, or alveolar epithelial cells.
Footnotes
Acknowledgements
For their invaluable necropsy and histology support, we wish to thank Mary Thiel, Kathy Henze, Ryan Vance, and June Wingert of Lexicon Pharmaceuticals. We also thank Dorothy Bush, Kimberly Robertson, and Pam Johnson of the Veterinary Pathology Core Laboratory of St. Jude Children’s Research Hospital for the excellent histochemical and immunohistochemical stains.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This research was funded by Lexicon Pharmaceuticals, Inc, The Woodlands, TX.