
Abstract
Hepatic encephalopathy has been listed as a differential for llamas displaying neurologic signs, but it has not been histopathologically described. This report details the neurologic histopathologic findings associated with 3 cases of hepatic lipidosis with concurrent neurologic signs and compares them to 3 cases of hepatic lipidosis in the absence of neurologic signs and 3 cases without hepatic lipidosis. Brain from all 3 llamas displaying neurologic signs contained Alzheimer type II cells, which were not detected in either subset of llamas without neurologic signs. Astrocytic immunohistochemical staining intensity for glial fibrillary acid protein was decreased in llamas with neurologic signs as compared to 2 of 3 llamas with hepatic lipidosis and without neurologic signs and to 2 of 3 llamas without hepatic lipidosis. Immunohistochemical staining for S100 did not vary between groups. These findings suggest that hepatic encephalopathy may be associated with hepatic lipidosis in llamas.
Keywords
Hepatic lipidosis has been reported with some frequency in llamas.6,16,17,19 The most common clinical signs include anorexia and weight loss. Biochemical abnormalities include elevated serum bile acids, γ-glutamyl transferase, aspartate aminotransferase, nonesterified fatty acids, and β-hydroxybutyrate, as well as hypoproteinemia, occasional hyperglycemia, occasional hypertriglyceridemia, and elevated sorbitol dehydrogenase.6,16,17,19 A feed deprivation study in llamas also revealed elevations in plasma insulin, triglyceride, and nonesterified fatty acid concentrations. 8
The pathogenesis of hepatic lipidosis in the camelid is poorly understood, with protein-energy malnutrition thought to be a primary contributing factor. Two common causes of protein-energy malnutrition are pregnancy and lacation,17,18 suggesting similarity to the pathogenesis of pregnancy ketosis in ruminants. 6 Extreme heat or cold stress18,19 and poor quality forage 5 may also result in protein-energy malnutrition. Presumptively, an increase in serum cortisol and decrease in insulin:cortisol ratio would follow environmental stressors.6,16,17 Additional studies suggest that the stress effect may be associated with elevated catecholamine levels.7,8 Inflammation may contribute to protein-energy malnutrition via inhibition of lipoprotein lipase and subsequent hyperlipemia. 6 Hyperlipemia and hypertriglyceridemia may occur alone or with other indicators of negative energy balance. 20
Primary or secondary alterations in insulin production or glucose metabolism may also contribute to the development of hepatic lipidosis. Hyperglycemia is considered relatively common in nonpregnant, nonlactating camelids with hepatic lipidosis.3,6
Hepatic Encephalopathy
This report presents 3 cases of hepatic encephalopathy diagnosed in llamas submitted to the University of Tennessee College of Veterinary Medicine for postmortem examination between 2006 and 2009. Hepatic encephalopathy has been suggested as a differential for neurologic disorders in llamas2,13,21 but, to our knowledge, has never been directly reported. Neurologic signs have been reported in llamas with hepatic lipidosis. 17 In other domestic species, hepatic encephalopathy is characterized by neurologic signs in association with acute or chronic hepatic disease.12,15 Histopathologic lesions typically include swollen astrocytes with large vesicular nuclei and prominent nucleoli, as well as cerebral edema or spongiosis.1,9
The pathogenesis of hepatic encephalopathy is incompletely understood. Ammonia, considered to be the primary toxin,12,15 is predominantly dietary in origin. 12 In hepatic dysfunction, ammonia may bypass the liver and diffuse into the brain via the blood or cerebrospinal fluid. 12 Ammonia is taken up by astrocyte and converted to glutamine by glutamate synthetase. 12 Accumulation of excess glutamine within astrocytes may result in osmotic stress and astrocytic swelling 12 via induction of mitochondrial permeability transition. 11 According to the “Trojan horse” hypothesis, accumulated glutamine may serve as a carrier for additional ammonia. 1
Histologic Methods
Cases were selected for this retrospective study on the basis of histopathologic lesions. Three llamas with histopathologic evidence of hepatic lipidosis and hepatic encephalopathy were identified in the tissues archived between 2006 and 2009 at the University of Tennessee College of Veterinary Medicine. A review of all archived llama cases from the same period was performed. The 3 most recent cases with histopathologic evidence of hepatic lipidosis and without evidence of significant encephalic lesions and the 3 most recent cases without histopathologic evidence of significant hepatic or encephalic lesions were selected for comparative purposes. Medical records stored within the University of Tennessee College of Veterinary Medicine for each of the 9 selected llamas were reviewed. Sections from all specimens were stained with hematoxylin and eosin. Immunohistochemical staining of cerebral tissue for glial fibrillary acid protein and S100 protein was performed using Zymed rabbit glial fibrillary acid protein primary antibody at a dilution of 1:400 for 30 minutes and Dako rabbit S100 protein primary antibody at a dilution of 1:10,000 for 30 minutes. Slides were labeled via the Dako labeled polymer system, using EnVision plus system HRP anti-rabbit, for 30 minutes. Slides were then stained with Dako plus diaminobenzidine and counterstained with hematoxylin. Samples were coded and evaluated blindly for intensity of astrocytic staining with each marker by both authors. The intensity scale was set as follows: 0 = no appreciable astrocytic staining, 1 = pale staining, 2 = moderate staining, 3 = dark staining.
Results
Signalment, clinical signs, antemortem bloodwork, and type of death are summarized in Table 1. On gross and microscopic examination, llamas Nos. 1–6 exhibited varied degrees of hepatic lipidosis. Liver sections from llama Nos. 7 and 9 were within normal limits, with mild hepatic fibrosis in liver sections from llama No. 8. In 3 cases, cerebral (llamas Nos. 1–3), hippocampal (llama Nos. 2 and 3), thalamic (llama No. 3), and cerebellar (llama No. 3) Alzheimer type II astrocytosis was characterized by single or clustered astrocytes with enlarged, vesiculate nuclei and inconspicuous cytoplasm (Fig. 1). Cerebral sections from llama Nos. 4, 5, and 7–9 were within normal limits, with focal meningeal mineralization and slight gray matter gliosis evident in cerebral sections from llama No. 6. Glial fibrillary acid protein and S100 protein staining intensity scores for individual llamas are presented in Table 2. Staining intensity for glial fibrillary acid protein was decreased in llamas with neurologic signs (Fig. 2) as compared to 2 of 3 llamas with hepatic lipidosis and without neurologic signs (Fig. 3) and 2 of 3 llamas without hepatic lipidosis (Fig 4). Immunohistochemical staining for S100 did not vary between groups (Figs. 5–7).
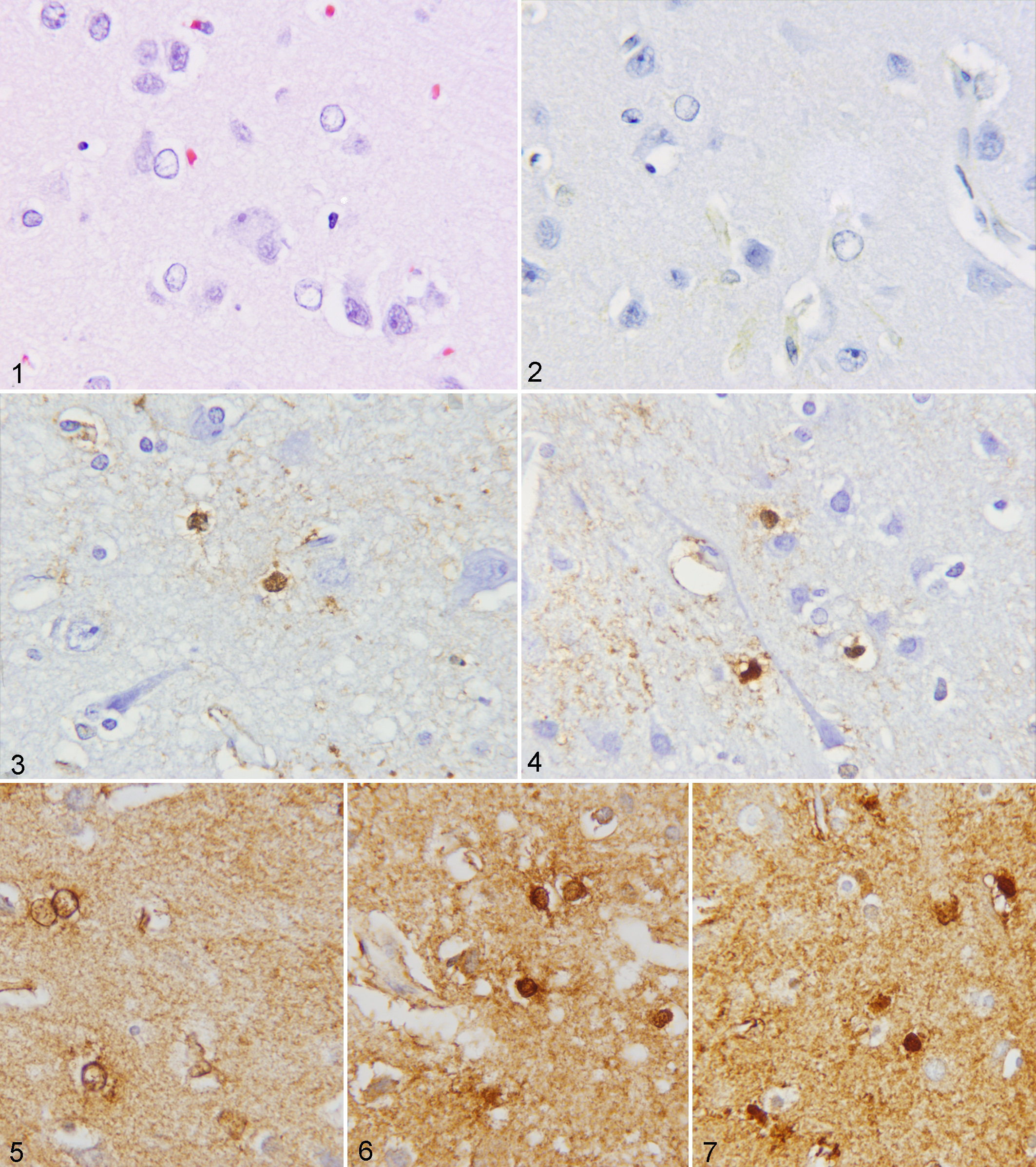
Signalment and Clinical Findings of Llamas With and Without Hepatic Encephalopathy and Lipidosis.a

a Llama Nos. 1–3, hepatic encephalopathy and hepatic lipidosis; llama Nos. 4–6, hepatic lipidosis only; llama Nos. 7–9, without encephalopathy or lipidosis. AST, aspartate aminotransferase; GGT, gamma glutamyl transpeptidase. All females were nonpregnant and nonlactating.
Subjective Staining Intensities for Glial Fibrillary Acid Protein and S100 Protein in Llamas With and Without Hepatic Encephalopathy and Lipidosis.
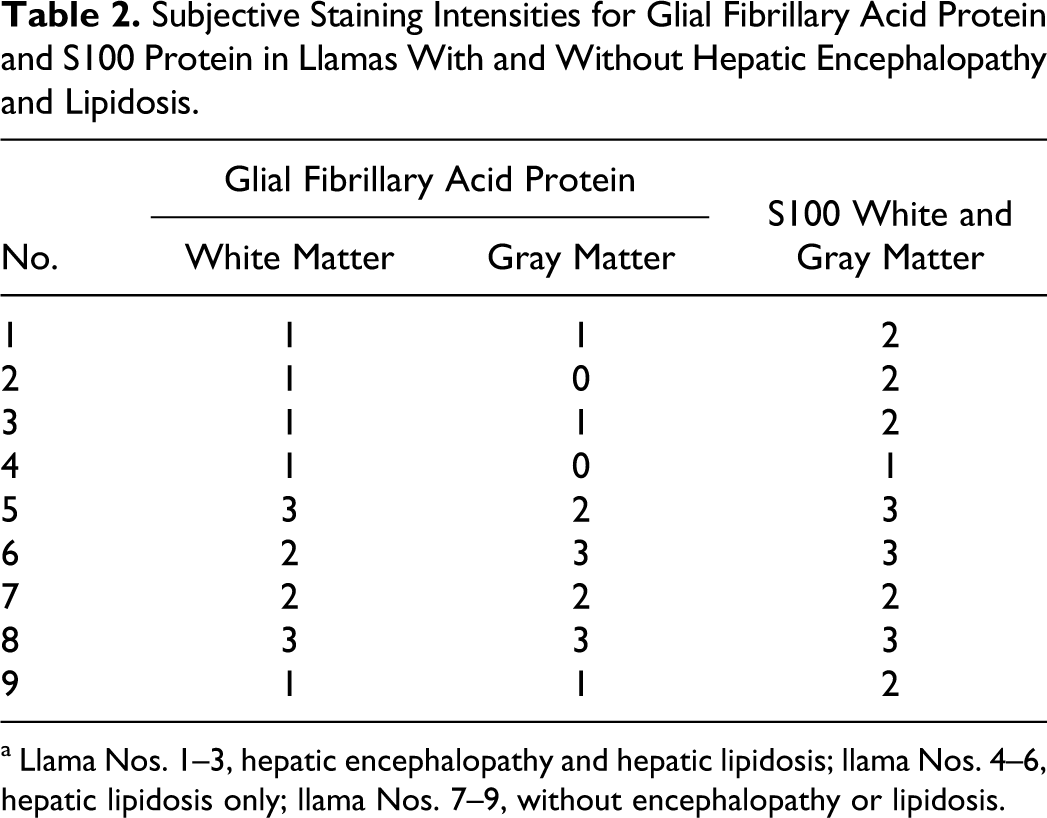
a Llama Nos. 1–3, hepatic encephalopathy and hepatic lipidosis; llama Nos. 4–6, hepatic lipidosis only; llama Nos. 7–9, without encephalopathy or lipidosis.
Discussion
Neurologic signs reported in the llamas identified in this study are summarized in Table 1. The neurologic signs reported in this study are consistent with those previously reported in llamas diagnosed with hepatic lipidosis 17 and suspected to be associated with hepatic encephalopathy in horses, 10 cattle, 4 and sheep. 14
Bloodwork was available for only 1 llama with clinical and histopathologic evidence of hepatic encephalopathy. Findings are reported in Table 1. Horses with histopathologic evidence of hepatic encephalopathy have been reported to have elevated serum ammonia levels, 10 which were not available for the cases in this study.
Alzheimer type II cells were identified in brain sections taken from 3 of the 9 llamas reviewed. This finding is consistent with the Alzheimer type II astrocytosis reported in horses diagnosed with hepatic encephalopathy 10 but inconsistent with the status spongiosus reported in cattle and sheep. 14
Llamas diagnosed with hepatic encephalopathy in this study displayed decreased subjective astrocytic staining intensity for glial fibrillary acid protein in both gray and white matter, with no change in S100 staining, as compared with llamas diagnosed with hepatic lipidosis–only and control llamas. A single case diagnosed with hepatic lipidosis and a single control case also displayed decreased staining for glial fibrillary acid protein in both gray and white matter; however, tissue preservation and, therefore, slide quality were poor in both cases, potentially resulting in spurious intensity scoring. Our findings are consistent with staining patterns for glial fibrillary acid protein and S100 protein reported in horses with idiopathic hyperammonemia. 10
Additional studies including serum blood ammonia levels and more thorough and systematic examination of brain sections would potentially allow for statistical evaluation of lesion predominance and distribution and for correlations among signalment, clinical signs, biochemical abnormalities, underlying disease, and histopathologic lesions. Transmission electron microscopy and development of an immunohistochemical protocol for glutamate synthetase in llamas may allow for confirmation of glutamine accumulation within llama Alzheimer type II cells. Finally, the effects of treatment of anorectic llamas displaying neurologic signs for ammonia toxicity and/or cerebral edema warrant investigation.
Footnotes
Acknowledgements
We acknowledge David Durtschi, Dr Shelley Newman, and the University of Tennessee College of Veterinary Medicine Histopathology Laboratory for their assistance in staining and immunohistochemistry.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
Funded by the University of Tennessee College of Veterinary Medicine Department of Biomedical and Diagnostic Sciences resident research allocation fund.