
Abstract
The high incidence of mammary tumor disease reported in certain canine breeds suggests a significant genetic component, as has already been described in human familial breast cancer—in BRCA1- and BRCA2-associated breast cancer in particular. The identification of genetic risk factors is critical to improvements in the prevention, diagnosis, and treatment of these tumors. In recent years, there has been significant progress in developing the tools and reagents necessary to analyze the canine genome. This work has culminated in a high-quality draft genome sequence, as well as a single-nucleotide polymorphism map and single-nucleotide polymorphism arrays for genomewide association analysis. These tools provide an unprecedented opportunity to characterize the genetic influences in canine diseases such as cancer, eventually allowing for exploration of more effective therapies. Given the high homology between the canine genome sequence and its human counterpart—as well as the many similarities regarding the morphology, biological behavior, and clinical course of mammary tumors in both species—the dog has proven to be an excellent comparative model. This review highlights the comparative aspects regarding certain areas within molecular biology, and it discusses future perspectives. The findings in larger genomewide association analyses and cDNA expression arrays are described, and the BRCA1/BRCA2 complex is compared in detail between the 2 species.
The spontaneous occurrence of canine mammary tumors (CMTs) has long been claimed to provide a suitable model for human breast cancer (BC) in a number of ways. The unraveling of the complete canine genome sequence in 2005 has finally permitted investigation of the molecular biological similarities in human and canine tumor entities. 40
For this review, we focused on the powerful new multicenter genomewide association studies (GWAS) of human BC and the lessons learned from those studies regarding the benefits and pitfalls, such as liability, clinical benefit, patient security, basic science on cancerogenesis, and economic rationale. Although no GWAS have yet been performed in dogs, it is only a matter of time until they are, given that dog-specific genomewide arrays are already available and have been shown to work for several disease phenotypes. 25,61 In the review, we also sought to couple the changes seen in germline mutation with the whole genome cDNA expression arrays. Because it is impossible to cover all aspects of molecular biology in mammary tumors both human and canine, we elected to discuss the findings from the larger contemporary studies, the similarities found in the 2 species, and finally, the likely next steps in the GWAS era of comparative mammary tumor oncology.
Human BC
It has long been known that BC is more common within certain families; hence, a genetic predisposition can be anticipated. The same is true for dogs, wherein the prevalence of mammary tumors is much higher in some breeds. 13 BC is actually twice as common in the first-degree relatives of women who have the disease as in the general population. 8 Although 2 major susceptibility BC genes, BRCA1 and BRCA2, were detected during the 1990s, up to 75% of cases in families with a high incidence of BC cannot be explained by the known BC loci. 12
Identifying risk factors for tumor development in general is needed to reduce mortality, select groups for screening programs, and increase the public’s awareness of the risks. The same rationale applies for BC, where known risk factors are considered, such as age at menarche, age at first live birth, postmenopausal hormone therapy, obesity, alcohol consumption, certain benign breast conditions, and presence of first-degree relatives with BC. When the Human Genome Project finished the sequencing of the human genome in 2001, 39,72 hope was high that it would lead to major breakthroughs in understanding oncogenesis in, for example, BC. 17
Much focus has been put on building up large biobanks of genomic DNA and tumor tissue from patients with BC, and many studies have been published on susceptibility loci and candidate genes for increased BC risk. However, the human genome is complicated to use, and the diversity in patients' lifestyles, geographic location, and other factors makes the findings somewhat nonconforming; thus, there is clearly a need for other spontaneous tumor models. In a 2009 study, Gail added 7 genotypes from single-nucleotide polymorphisms (SNP), known to be risk factors for BC, to an already-validated BC risk model—the National Cancer Institute’s BC Risk Assessment Tool. The researcher found that doing so increased the ability to improve the expected number of life-threatening events by only 0.81% and 0.07% for women aged 50 to 59 and 40 to 49 years, respectively, in terms of deciding whether to take tamoxifen to prevent BC. 18
Cancer diagnostic research does not differ from research in other disciplines in the continuing search for greater understanding and resolution of disease mechanisms. In molecular biology, that means migrating from simply recognizing the presence of genetic association in BC to investigating microsatellites and detailed candidate gene approaches at the chromosome level and, finally, to attacking the complicated multifactorial aspects of BC development with powerful GWAS SNP analyses, combining the germline genetic changes with several thousand genes in cDNA microarrays at the tumor tissue level and thereby defining gene expression in the cancer itself.
Known BC Susceptibility Genes in Humans
BRCA1 and BRCA2 genes
In the early 1990s, researchers discovered a breast and ovarian cancer susceptibility gene on choromosome 17 using positional cloning methods. 44 The gene was called BRCA1. The authors concluded that identification of BRCA1 should facilitate early diagnosis of breast and ovarian cancer susceptibility in some individuals, as well as a better understanding of BC biology. The same year, Wooster et al described the second BC susceptibility gene, BRCA2, in 15 high-risk BC families, showing the gene to have no link to BRCA1. 81,82 BRCA2 was localized to chromosome 13, as found through linkage analysis. 82
Together, BRCA1 and BRCA2 are thought to act as tumor suppressors only. Hence, mutations in these genes will lead to the cells' decreased ability to repair DNA damage, such as DNA double-strand breaks. DNA damage accumulates over time, and new mutations and dysfunctions propagate, with the cells becoming more prone to develop into cancer cells. In the case of BRCA1 and BRCA2, the cell injuries will occur in tissues where the protein is expressed. Indeed, many cancers—breast, ovarian, fallopian tube, prostate, and pancreatic cancers, as well as malignant melanomas—are associated with mutations in BRCA1 and BRCA2.
A specific type of anemia, Fanconi anemia, is present if 2 defect copies of BRCA2 are inherited. 41 In this syndrome, patients are prone to develop certain types of leukemia and lymphomas; solid tumors found preferentially in the head and neck, further indicating the BRCA pathway’s central role in tumor suppression and DNA repair. 47 There is not the scope to examine the syndrome in this review, but Fanconi anemia is one of many examples where malfunctions in DNA repair genes give rise to a wide spectrum of tumors.
Although BRCA1 and BRCA2 mutations are shown to be present in families with a high incidence of BC, several studies have proven that in fact only a small proportion of the families carry the mutations; hence the mutations cannot explain the majority of the cases of BC. In a study in 1999, Peto et al 53 concluded that the respective percentages of BRCA1 and BRCA2 mutation carriers are 3.1% and 3.0% of BC patients below 50 years of age, 0.49% and 0.84% of BC patients 50 years of age and above, and 0.11% and 0.12% of women in the general population.
BRCA1 and BRCA2 made approximately equal contributions to early-onset BC in Great Britain and accounted for a small proportion of the familial risk of BC. 53 A later large meta-analysis study based on 22 reports confirmed the findings of Peto et al, 3 with the authors concluding that the average risks of BC and ovarian cancer are both higher in BRCA1 mutation carriers than in BRCA2 mutation carriers; however, the difference decreases for BC after 50 years of age. The relative risk of BC in BRCA1 mutation carriers, compared to general population rates, declines with age, from greater than 30-fold at < 40 years of age to 14-fold at > 60 years of age; in contrast, the relative risk in BRCA2 mutation carriers is approximately 11-fold in all age groups at > 40 years of age and is not significantly higher at earlier ages. As a result, the incidence rates in BRCA1 mutation carriers rise to a plateau of approximately 3 to 4% per annum in the 40- to 49-year age group and are roughly constant thereafter, whereas the BRCA2 rates show a pattern similar to those in the general population, rising steeply up to age 50 and more slowly thereafter.
There is also a reported difference in cancer risk depending on where the BRCA mutations are located. For BRCA2, the risk of BC is lower if the mutation is situated in the ovarian cancer cluster region (where the risk of developing ovarian cancer is significantly increased), whereas for BRCA1, the risk of BC is lower if the mutation is found in the central part of the gene. 3 In conclusion, the BRCA complex has failed to explain the majority of familial BC cases (about 75%) and accounts for only a tiny percentage of sporadic nonfamilial BC (about 5%).
An important alternative route of genetic regulation is epigenetic control, mostly due to hypomethylation/hypermethylation of the DNA. In humans, BRCA1 has been found to partly be controlled by, for example, promoter methylation, which explains sporadic BC even where no mutations are present. 10,67 The epigenetic regulation of canine cancers is poorly covered and will not be discussed this article, simply due to lack of data. In a recent article by Vinothini et al, epigenetic regulators such as DNA methyltransferase (Dnmt-1) and histone deacetylase (HDAC-1) were investigated in 30 mammary tumors. 73 The study was small, and findings must be reproduced before the epigenetic regulation in CMTs can be reviewed in a meaningful way.
Finally, BRCA2 is reported to be modulated by expression of negative regulators such as EMSY, which binds to exon 3 of BRCA2 and suppresses activation of a reporter construct by a BRCA2–GAL4 fusion protein and hereby also nonmutated BRCA2 sporadic BCs be partly explained. 22
Other BC: Susceptibility genes in humans
Following the notion that BRCA1 and BRCA2 mutations do not explain the majority of BC cases in hereditary BC syndrome, large studies on genetic linkages have attempted to suggest other genes. The complexity of the human genome and the high heterogeneity within non-BRCA1/BRCA2 families are often emphasized in these trials. In one of the first GWAS using SNP markers to identify BC susceptibility loci, Gonzales-Neira et al used a 4.720-SNP array (Illumina Linkage III Panel, Illumina, Inc, San Diego, CA) to scan across the genome in 19 non-BRCA1/BRCA2 families (a total of 82 cases). 19 The study reported 6 regions on chromosomes 2, 3, 4, 7, 11, and 14 as candidates to contain genes involved in BC, with 5 of these regions confirmed in further fine mapping. The authors concluded that the 2-stage linkage mapping strategy was optimal but needed to improve in power. They recommended including large subsets of families from homogeneous populations in future studies, to reduce the genetic variation and increase the linkage detection power. Moreover, the authors wrote that the density of the array needed improving to give higher resolution and reduce the size of the susceptibility loci. The current choice of multi-SNP arrays still leaves scientists with regions of the genome containing 300 genes that show higher risk in BC families. 4,59
Some of the other genes that researchers believe contribute to a higher risk of BC are checkpoint kinase 2 (CHEK2), fibroblast growth factor 2 (FGR2), transformation-related protein 53 (TP53), and phosphatase and tensin homolog (PTEN). 76 Most of these genes are involved in cancerogenesis as DNA repair genes—and, hence, also tumor suppressor genes much like BRCA1 and BRCA2. Recent reports have, however, described genes that are involved in the cell proliferation pathways as well: FGFR2, RAD50 (RAD50 homolog [Saccharomyces cerevisiae]), mitogen-activated protein kinase 1 (MAP3K1), which also has a metabolic role, and transforming growth factor β 1 (TGFB1), which is also involved in differentiation. 9,12,23,64
To summarize, BC susceptibility genes can be grouped by whether they make a high-, intermediate-, or low-risk contribution to tumor development. 56 The highest risk is seen in BRCA1 and BRCA2, followed by cadherin 1 (CDH1), nibrin (NBH), neurofibromin (NF1), PTEN, TP53, and serine/threonine protein kinase 11 (STK11). In the intermediate group, genes such as ataxia-telangiectasia mutated (ATM), BRCA1 interacting protein C-terminal helicase 1 (BRIP1), CHEK2, partner and localizer of BRCA2 (PALB2), and RAD50 are found. In the group of genes contributing the least to known increases in BC risk are FGFR2, lymphocyte-specific protein 1 (LSP1), MAP3K1, TGFB1, and TOX high mobility group box family member 3 (TOX3).
Because many of these genes are general tumor suppressors or are involved in cell proliferation and differentiation processes, disturbances in these genes give rise to not only increased BC risk but also other tumor types. Some examples are ovarian cell carcinoma (BRCA1 and BRCA2); soft tissue sarcomas, leukemia, and brain tumors (TP53; see Li-Fraumeni syndrome, eg); diffuse gastric cancer (CDH1 or cluster of differentiation 324 [CD324]); and colorectal cancer (mutS homolog 2 [MSH2] protein). 28,56,62,71
Unfortunately, the current ability of susceptibility genes to contribute to the overall risk analysis is still poor. In a recent article, Wacholder et al described a population of 5,590 BC subjects and 5,998 controls, 50 to 79 years of age, from 5 studies (4 in the United States and one in Poland). They concluded that the addition of 10 common genetic variants associated with BC only modestly improved the performance of risk models for BC—including information on age, study, and entry year and 4 traditional risk factors of age at menarche, number of biopsies, age at first live birth, and number of first-degree relatives with BC. 75 The area under the curve was 61.8% when the 10 genetic variants were added to the model, compared with 58.0% before their addition.
Future tasks are to refine analysis, select (large) cohorts more uniformly, and, finally, look for help from spontaneous mammary tumor models in other species, such as the dog.
Gene Expression Studies in Humans
It has long been recognized that the expression of sex hormone receptors and human epidermal growth factor receptor 2 (HER2) is important in describing clinical behavior, treatment, and prognosis. The past decade has seen the introduction of more refined tools to examine BC gene expression, mainly based on cDNA microarrays. The advantage of these tools is that they permit simultaneous and less biased investigation of thousands of genes, compared with the examination of a single gene or several genes at a time using reverse transcription polymerase chain reaction methods or immunohistochemical analyses. The disadvantages of mRNA expression analyses are that, first, the information yielded does not indicate if the altered expression is the result of, for example, mutations, gene copy number changes, or regulatory gene changes. Second, mRNA investigation does not always reflect the final outcome of protein expression, given that posttranslational modifications are not studied. Another problem with gene expression profiling is that most oligos are not optimized (this is, of course, also true for canine expression analyses). They may not be very sensitive for a particular gene; hence, moderate differences in expression of some genes may not be picked up, although they are relevant. Moreover, microarrays do not usually discriminate between splicing variants. Accepting these pitfalls, cDNA microarrays are still extremely powerful and have been shown to reproduce results in many independent studies in recent years.
The detailed characterization of BCs strives to establish better prognostic and predictive signatures: Prognostic signatures are useful to decide who should be treated with chemotherapy, given that patients with a good prognosis should not suffer from toxic drugs. Prognostic signatures flourish because they reflect tumor cell behavior that depends on the dysregulation of pathways—that is, many genes, mRNAs, and proteins.
Predictive signatures are used to decide what a patient should be treated with. Thus far, there is only the HER2 and hormone receptor status. Unfortunately, estrogen- or HER2-targeting therapies are not sufficient for such a heterogeneous disease as BC, given that many patients with metastatic estrogen receptor–positive (ER-positive) or HER2-positive tumors do not respond to targeted therapy only. Moreover, about 15% of all BCs are hormone receptor negative and HER2 negative. Unfortunately, there is no predictive signature that can be used in the clinic for conventional chemotherapy. However, it is also more difficult because small changes in only one of many proteins can cause drug resistance (eg, cAbl mutations and Gleevec resistance). 1 But such small differences are not easy (or impossible) to detect with gene expression profiling.
The first expression studies performed in early 2000 divided BC into 4 subtypes referred to as intrinsic because they are defined more by the tumors' intrinsic properties than by their behavior. 52 The 4 subtypes are basal-like, HER2 enriched, luminal A, and luminal B. 6 The first 2 subtypes are named for their expression of basal-like cluster genes (cytokeratins 5, 14, and 17; epidermal growth factor receptor [EGFR]; c-kit thyrosine kinase; vimentin; and P-cadherin) and high HER2, whereas the latter 2 luminal subtypes are expressing genes similar to those in the luminal epithelial component of the breast. Further tumor subcategorization is possible—for example, ER negative, progesterone receptor (PR) negative, and occasionally, triple negative (eg, ER, PR, and HER2). This subcategorization, of course, guides in use of different treatment modalities and, to some extent, prognosis. 11,51
It has recently been reported that the subtype classifications risk being poorly reproducible. When different single-sample predictors were tested, they did not reliably assign the same patients to the same molecular subtypes, except for the group of basal-like BC. 77 The authors stated, “For molecular subtype classification to be incorporated into routine clinical practice and treatment decision making, stringent standardization of methodologies and definitions for identification of BC molecular subtypes is needed.” This will be an important lesson for the veterinary field, given that new techniques are emerging to start molecular classification of CMTs as well.
A milestone in gene expression profiling was the 70-gene signature by van’t Veer et al. 70 It is now commercialized as MammaPrint and compared with other clinical parameters (MINDACT trial). Although this trial is ongoing, it is reported that the combination of clinical and molecular prognostic factors seems to be most useful for clinicians to decide whether patients with ER-positive, lymph node–negative BC should receive chemotherapy or not. 5
Before the discovery of HER2 and the development of HER2-targeted therapy with the monoclonal agent trastuzumab (Herceptin), the HER2-enriched tumor entity had a poor outcome. 63 Unfortunately, except for the ER antagonist tamoxifen 24 and aromatase inhibitors, few new discoveries have been made since Slamon and colleagues' work 63 that have even modestly affected treatment outcome based on knowledge of tumor gene expression patterns.
Basal-like BC also remains a challenge to be overcome. In this subtype, the sign of dependence of EGFR for growth and proliferation called for studies on anti-EGFR monoclonal antibodies. A study using cetuximab in combination with carboplatin showed an association with clinical response as EGFR expression was inactivated, but most tumors failed pathway inactivation despite the targeted drug therapy. 6 Basal-like tumors are also associated with BRCA1 mutation carriers and hence have a corrupted DNA repair system. Thus, trials have been initiated to target the poly(ADP-ribose) polymerase (PARP) pathway that constitutes a high-fidelity DNA repair pathway. If PARP also becomes disrupted, cells with BRCA1 dysfunction will eventually die. Orally administered PARP inhibitors have recently been shown to give a 63% response rate in a phase 1 trial in late-stage breast, ovarian, and prostate cancer patients with BRCA1 and BRCA2 mutations. 16
Despite these exciting and significant milestones in the detection of gene expression in BC, the Evaluation of Genomic Applications in Practice and Prevention Working Group recently found insufficient evidence to recommend either for or against the use of tumor gene expression profiles in improving outcomes in defined populations of women with BC. 14 The group’s statement reflects the complexity of this tumor category and the need for further investigation and increased understanding of tumor biology, tumor behavior, and new treatment modalities in BC—a disease that continues to strike so many women.
CMTs
The unraveling of the dog genome in 2005 set the starting point for many studies in comparative diseases, given that the genome was shown to have high homology with its human counterpart. 40 Because the mapping of the canine genome was completed several years after the mapping of the human genome, fewer data are available on genetic susceptibility to CMTs and the like. Nevertheless, the dog tumor model has many more advantages than do induced murine tumor systems, and dog owners largely want to improve breeding and thereby avoid obvious enrichment of tumors in certain breeds. Additional factors are the increasing awareness of companion animal welfare, ethical concerns over the use of laboratory animals, and the development of veterinary specialties such as oncology, with the volume of studies they entail constantly on the rise. 29,50
Direct genetic mapping strategies are in place for many cancers in dogs and are of comparative interest in humans. 26 The LUPA Consortium, funded by the European Commission’s Seventh Framework Programme, aims to find genes responsible for at least 18 diseases, including 4 cancers, in the coming years. The work will entail a pan-European effort in collecting samples and phenotyping, as well as compiling GWAS data from 8,000 dogs. Identifying the polygenetic traits of the cancers will, of course, pose a huge challenge, but given the breed structure, the genetic composition resulting from important bottlenecks (Fig. 1 ), and the high incidence of certain cancers within certain breeds, the number of cases and controls needed is estimated to be extremely small compared with the number for human studies. 26 This estimate is already shown to be accurate for monogenic and autosomal dominant traits. 25,61
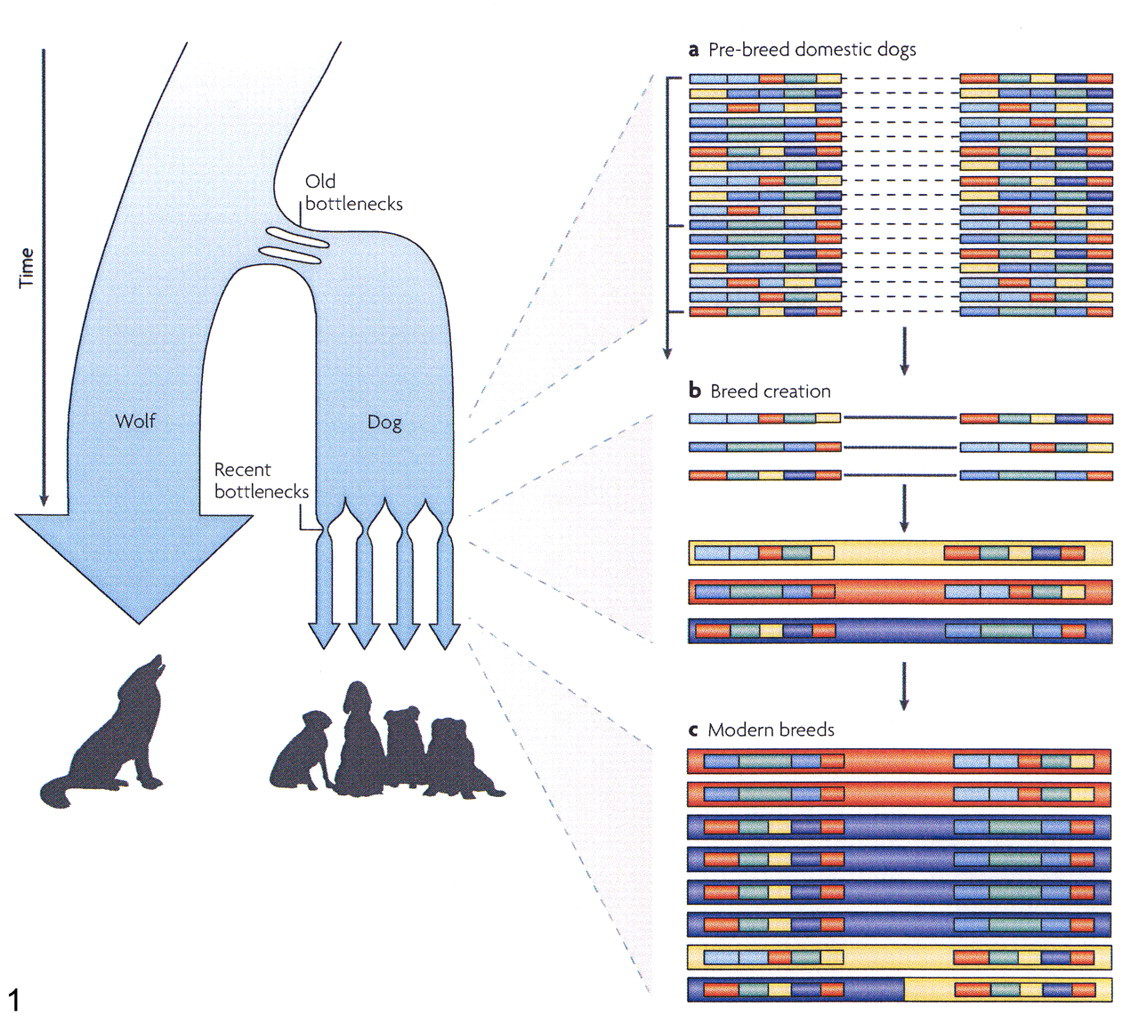
Haplotype structure of the dog. Two population bottlenecks in dog population history, one old and one recent, shaped haplotype structure in modern dog breeds. First, the domestic dog diverged from wolves about 15,000 years ago, probably through multiple domestication events. Within the past few hundred years, modern dog breeds were created. Both bottlenecks influenced the haplotype pattern and linkage disequilibrium (LD) of current breeds. (a) Before the creation of modern breeds, the dog population had the short-range LD that would be expected given its large size and the long period since the domestication bottleneck. (b) In the creation of modern breeds, a small subset of chromosomes was selected from the pool of domestic dogs. The long-range patterns that were carried on these chromosomes became common within the breed, thereby creating long-range LD. (c) In the short time since breed creation, these long-range patterns have not yet been substantially broken down by recombination. Long breed haplotypes, however, still retain the underlying short ancestral haplotype blocks from the domestic dog population, and these are revealed when one examines chromosomes across many breeds. This figure is modified with permission from Nature Review Genetics, 26 Macmillan Publishers Ltd. All rights reserved. We would also like to thank Drs Karlsson and Lindblad-Toh for this illustration.
Known Mammary Tumor: Susceptibility Genes in Dogs
BRCA1 and BRCA2 genes
The canine gene sequences for the BRCA1 and BRCA2 proteins have both been described. 49,65 Studies of germline mutations in the dog and association with mammary tumors are, however, very few. Most studies investigated gene expression in tumor tissue. A large candidate gene approach was recently presented by Rivera et al, who showed that germline mutations in BRCA1 and BRCA2 were associated with a significantly increased mammary tumor risk in a breed with known prevalence to mammary tumors in Sweden (Bonferroni-corrected P = .005 and P = .0001, respectively), resulting in both BRCA1 and BRCA2 conferring an approximate 4-fold increase in risk for CMT. 13,57
CMT expression of mutated BRCA1 and BRCA2 was reported in 2001 by Ochiai et al, who also discussed the coreaction between BRCA2 and RAD51. 49 RAD51 is a DNA-repairing protein shown to be involved in mediating mammary tumors if altered. 27 As with other studies in veterinary medicine, the true pitfall is the small sample numbers available in each study and the lack of information on pedigree. Nevertheless, a number of studies have similarly concluded that BRCA-ness seems to be an important feature of mammary tumor development in dogs.
In 2003, Nieto et al showed that BRCA1 expression was significantly reduced as malignancy developed in CMTs. 48 The study was performed on 44 dogs with neoplastic mammary gland tumors, 7 dogs with dysplastic mammary gland tumors, and 2 normal controls. The reduced immunohistochemical expression coincided with high proliferation marker Ki-67 and ER-α-negative tumors. 48
In 2010, Klopfleisch et al investigated differential mRNA expression of BRCA1, BRCA2, and RAD51 in laser-microdissected tissue samples (n = 16) of normal mammary gland epithelium, simple canine adenomas, and adenocarcinomas, thus avoiding contamination by stromal and nonneoplastic epithelial cells. 32 To investigate the expression, quantitative real-time polymerase chain reaction (qRT-PCR) was used. In this study, BRCA1 expression was not unequivocally associated with the histologic criteria of malignancy and therefore differed from human malignancy. RAD51 was overexpressed in 80% of lymph node metastases. Similarly, 50% of the primary tumors had RAD51 overexpression. Furthermore, BRCA2 and RAD51 were, in parallel, overexpressed in 5 of 10 lymph node metastases. The findings are somewhat conflicting, given that one might expect reduced expression instead increased suppression in the more malignant/transformed cells. The authors concluded that this overexpression could be due to a negative regulatory loop in which BRCA2 is induced by cellular proliferation, which then inhibits proliferation. 54 In any event, the findings are interesting and warrant further study in larger populations of tumors, with the germ-line BRCA1 and BRCA2 status defined as well.
In an accompanied study, the same group investigated RAD51 expression by immunohistochemistry in paraffin-embedded mammary carcinomas and their lymph node metastases (n = 40 dogs), adenomas (n = 48 dogs), and nonneoplastic mammary glands (n = 88 dogs). 37 RAD51 expression was increased in carcinomas and their metastases, indicating genomic instability. Paradoxally, the increased RAD51 overexpression was not confirmed by the qRT-PCR investigation in 2009 of the same condition. 32 These conflicting findings again show that cDNA studies do not automatically reflect the final outcome of protein expression because of posttranslational modifications.
In human studies, much focus has been on the BRCA2 region of exon 11 because this is the largest exon and it contains several BRC repeats binding to RAD51, which catalyzes homologous DNA pairing and DNA strand exchange, crucial for RAD51 interaction and the DNA repair mechanisms. 7,42 Recently, BRC repeats and RAD51 were investigated in the dog by Hsu et al. 21 The authors investigated 15 samples: 10 malignant, 1 benign, and 4 from normal controls. Several mutations (n = 19) were found, whereas many of these resulted in amino acid changes (68%), with 2 situated in the BRC repeating domain. 21 The authors concluded that there was an association between clinicopathologic status and the single-nucleotide variations in exon 11 of the BRCA2 gene, seemingly higher in cancerous mammary tissue than in healthy tissue. A high frequency of genetic variations in 2 “hot spots” (A511C and A2414G) was also seen in the majority of tumor types and tumor stages. Notably, both hot spots were observed in a dog with stage V mammary carcinosarcoma with a shorter survival time. Although it was a fairly small study, its authors ended by speculating that these hot spots may be a prognostic factor in dogs with malignant mammary tumors 21 —a speculation that, in our view, needs larger confirmatory studies before it can be accepted.
Other mammary tumor susceptibility genes in dogs
As previously mentioned, there is little information from larger studies about the existence, frequency, and importance on germline mutations and their role in the risk of developing mammary tumors in dogs. The lack of information is, of course, due to the relatively recent mapping of the canine genome sequence, and more details will become available as data are collected from larger cohorts of DNA from dogs with mammary tumors and negative controls. This collection is one of the main tasks of the LUPA Consortium initiative. In the study in 2009 where Rivera et al used a candidate gene approach on 10 known human BC susceptibility genes and where BRCA1 and BRCA2 were found to significantly contribute to the risk of developing mammary tumors in the English Springer Spaniel, the other selected genes failed to show an increased risk in this population. 57 The population consisted of 212 CMT cases and 143 controls. Apart from BRCA1 and BRCA2, the selected genes were CHEK2, ERBB2 (gene encoding HER2), FGFR2, LSP1, MAP3K1, RCAS1, TOX3, and TP53. A borderline association was seen for FGFR2 (best raw P value = .0047) but was lost after Bonferroni correction (P = .18).
Besides the Rivera et al study, no larger CMT studies with results on germline increased-risk mutations are currently available.
Gene Expression Studies in Dogs
In contrast to the germline mutation investigations, tumor expression studies in CMT are more abundant. The reason is that many studies have been performed on one or a couple of candidate genes, with known sequence, and adopted from already susceptibility human BC gene studies. Immunohistochemical expression studies are described elsewhere, and in this review, we consider mRNA expression studies only.
Candidate gene expression studies
A number of interesting expression studies in CMT have come from Dr Robert Klopfleisch and the group at the Department of Veterinary Pathology at the Freie Universität in Berlin. 30 –38 The studies have mostly employed a “few-gene” candidate gene approach, have been based on large samples, and have often been correlated to protein expression in confirmatory immunohistochemical studies. In addition, tumor samples have been collected with laser microdissection, giving highly representative tumor tissue samples and avoiding expression biases from, for example, normal cells and stromal tissue.
The Berlin researchers have reported reduced transcription of the stanniocalcin-1 gene and overexpression of the derlin-1 gene associated with malignant behavior (preferentially in metastases) in mammary adenocarcinomas. 30 Regarding the TP53-regulated cyclin-dependent kinase inhibitors p21 and p27, important for controlling cell cycle progression, it was found that loss of p21 overexpression was associated with tumor metastasis whereas reduced cell cycle inhibition by p27 was associated with malignant progression.
The transforming growth factor β (TGFβ) family was investigated regarding its role in the carcinogenesis of CMT in 2009. 35 When compared with nonneoplastic glands or adenomas, TGFβ3, TGFβR3, and latent transforming growth factor binding protein B4 (LTBP4), expression was significantly decreased in carcinomas and metastases whereas LTBP1 expression was increased. The mRNA expression pattern was confirmed at the protein level by immunohistochemical analysis of tumor tissue for TGFβ and LTBP4. The authors stated that the loss of TGFβ3 and LTBP4 may have growth-stimulating effects in late-stage tumors and that loss of their expression, together with reduced TGFβR3 expression, may be associated with increased proliferative activity of CMT, similar to findings in human BC. 35
Cell adhesion is an important regulator of cell growth and motility, such as proneness for tumor cells to metastasize. The recently described cell adhesion molecules 1 and 2 (HEPACAM1 and HEPACAM2) were investigated for their role in CMTs. 33 HEPACAM1 is involved in negative cell cycle regulation via TP53, p21, and p27 signaling; it also mediates increased human BC cell spread, whereas HEPACAM2 activity has still not been readily investigated. 45 The findings by Klopfleisch et al were not entirely expected, but the authors concluded that HEPACAM1 and HEPACAM2 seem to be important for cell–cell adhesion of normal and neoplastic canine mammary cells. However, they questioned the role of HEPACAM1 as a tumor suppressor at late stages of malignant transformation owing to loss of protein expression in adenomas but not in carcinomas, and they indicated that HEPACAM1 might rather be involved in physiologic mammary cell adhesion and CMT metastasis. 33
In a study with a larger set of markers (n = 49), Klopfleisch et al recently investigated expression patterns in a qRT-PCR assay in 10 benign and 13 metastatic CMTs. 34 The authors found 17 genes with significant (P < .05) differences in gene expression levels between benign and malignant tumors—including ERBB1, slit homolog 2 protein (SLIT2), PR, MIG6, special AT-rich sequence-binding protein 1 (SATB1), and SMAD6 (homolog of the Drosophila gene “mothers against decapentaplegic” / member of the TGFβ superfamily). However, none of them could be used alone to predict whether the tumor was benign or malignant. Used in combination, bone morphogenetic protein 2 (BMP2), LTBP4, and derlin-1 (DERL1) correctly classified each individual tumor as benign or malignant, pointing toward a possible first step in using molecular phenotyping as an aid in predicting malignant behavior in CMTs, as is already being done in human BC. 34
Mammary tumor expression in a number of mammals, including rats, humans, and dogs, have been studied in depth by the group at the Department of Comparative Pathology at the Veterinary School of the University of Cordoba in Spain, as led by Professor Juana Martin de las Mulas. However, most of the group’s publications refer to a range of immunohistochemical markers, and it is beyond the scope of this review to cover them all. In 2003, Martin de las Mulas et al described expression of the oncogene HER2 in canine mammary gland carcinomas, regarding both protein expression and correlation to gene expression measured by chromogenic in situ hybridization. 43 The authors found a protein expression of approximately 20%, reflecting the HER2 positivity reported in human BC. The in situ hybridization failed to detect any gene amplification, suggesting that the canine mammary carcinomas could serve as a good model for the subgroup of protein-positive HER2 human BCs that also lack detectable gene expression of this mammary oncogene. 43
In a study on 79 CMTs (32 benign and 47 malignant), Rungsipipat et al reported a fairly high frequency of HER2 expression in benign lesions (50%), as investigated immunohistochemically using antibodies against human c-erbB-2 (HER2) products. 60 These findings were not confirmed in a recent study on a larger case population by Hsu et al. 20 In this, 91 dogs with malignant mammary tumors, 6 with benign lesions, and 2 normal controls were used. HER2 protein overexpression was found in 27 of 91 (29.7%) canine malignant mammary tumors and none in the benign lesions. The dogs overexpressing HER2 also tended to have a better 2-year survival. Collectively, the results mimic the findings in human studies, and the authors concluded that the differences compared to earlier canine studies on HER2 expression could be due to a larger number of dogs included in their study (91 cases). Furthermore, the percentage of HER2 protein overexpression in the Hsu et al study was lower than the mRNA level of HER2 (74%) in late-stage CMTs described earlier. 2 HER2 overexpression may depend on a number of factors, including the sensitivity of detection methods, the level of gene expression (amplification, transcription, translation), or the stages of tumor samples.
In 2010 Ferreira et al described comparative aspects on columnar cell lesions (precancerous and neoplastic) in canine mammary gland specimens with the expression in similar lesions in human BC. 15 Columnar cell lesions were identified in 67 (53.1%) of the 126 canine mammary glands with intraepithelial alterations (epithelial hyperplasia and in situ carcinoma and invasive lesions). The authors investigated, among other markers, HER2 expression and found that it was negative in the canine changes (ER, PgR, and E-cadherin positive but negative for cytokeratin 34βE-12 and P53). The authors concluded that columnar cell lesions in canine mammary gland were hence pathologically and immunophenotypically similar to those in human breast. 15
Multimarker gene expression studies
The first multimarker (n = 174) cDNA expression analysis of CMTs was likely the study in 2005 by von Euler et al, who used 2 human multimarker cDNA microarrays: a human signal transduction pathway finder gene array with 87 genes and a human cancer pathway gene array with 87 genes. 74 The choice to use human arrays was due to a lack of canine cDNA arrays at the time and the suggested high homology of the coding sequences of the genes. In addition, the primary aim of the study was to elucidate the necessity of correct tissue handling adjacent to surgery and thereby achieve mRNA of proper quality to perform reliable expression analyses, given that the results of expression in the same tumor will differ significantly depending on where the samples are collected and if mRNA degradation has occurred. The expression in CMTs showed good correlation with what had been found in human BC and pathways, such as adhesion (intercellular adhesion molecule 1 [ICAM-1]), angiogenesis (angiopoietin-2 and IFN-β1), apoptosis (telomerase reverse transcriptase [TCS1]), cell cycle control and DNA damage repair (BRCA1, p21Waf1/CIP1 cyclin-dependent kinase inhibitor 1A [p21Cip1]), signal transduction molecules and protein transcription factors (RAS p21 protein activator [RasGAP]), and survival pathway (BCL2-related protein A1 [BFL1] and BCL2-like 1 protein [Bcl-x]). 74
Given that the whole genome canine expression arrays are now available, it is expected that the data will expand rapidly. No attempt has yet been made to classify CMTs into intrinsic subtypes, as has been done in human BC. The first true multimarker microarray on canine mammary cell lines was likely the study published in 2008 by Rao et al. 55 This cDNA array was based on a collection of approximately 21,000 canine genes. Three well-characterized cell lines were used: a canine mammary osteosarcoma (CMT-U335), a canine mammary atypical benign mixed tumor (CMT-U229), and P114 from a highly malignant canine anaplastic carcinoma. For detailed information on the different genes and their expression, see the original source.
In summary, the Rao study showed that in CMT-U229, many key cell cycle–activating cyclins (CCND1, CCNG1, and CCNI) were overexpressed and a cell cycle inhibitor (CDKN1A) was underexpressed. Overexpression in genes involved in cytokine/Rho-GTPase signaling (SOCS5, PAK1, and MYLK) and integrins ITGA6 and ITGB4 was also found in this atypical benign mixed tumor. Cell line P114 had high expression of many activators and direct targets of the Wnt pathway (CSNK1A1, CTNNB1, CCND1, DDR1, and OPN), plus lower levels of inhibitors of the Wnt pathway (DKK3, SFRP1). P114 also overexpressed the cell cycle inhibitor CDKN1A, laminins (LAMA3 and LAMB1), and fibronectin (FN1). The slowest-growing cell line, CMT-U335, revealed a large number of differentially expressed genes compared with P114 and CMT-U229, indicating that it is more distinct from the other 2 cell lines. This aggressive primary osteosarcoma overexpressed many genes involved in integrin signaling, such as collagens (COL12A1, COL1A2, and COL4A1), OGN, RAP1B, VCL, ITGB5, and MAPK14. 55
In 2009, 2 studies were published whose authors had looked at tumor tissue samples from 22 cases in an array covering more than 20,000 genes 78 and 33 samples analyzed by an array covering approximately 30,000 genes (Fig. 2 ). 68 In both studies, the different malignancy grades grouped rather appropriately. In the Wensman et al study, 78 the focus was on the specific pattern of expression in the sarcoma phenotype, already indicated in the in vitro study by Rao et al the year before. 55 Wensman et al found a specific expression of the sarcomas; in particular, the osteosarcomas showed a striking upregulation of a panel of homeobox genes previously linked to craniofacial bone formation. This finding was confirmed at the protein expression level by immunohistochemistry on tumor samples and in vitro after stimulating an osteosarcoma cell line with a bone morphogenetic protein (BMP-2). 78 The BMPs and genes associated with retinoic acid signaling were accordingly upregulated in the canine sarcoma mammary tumors. Not surprising, in the carcinomas, several genes involved in cell-to-cell adhesion and several genes related to epithelial differentiation—such as various cytokeratins, E-cadherins, and occludin—were all expressed at higher levels.
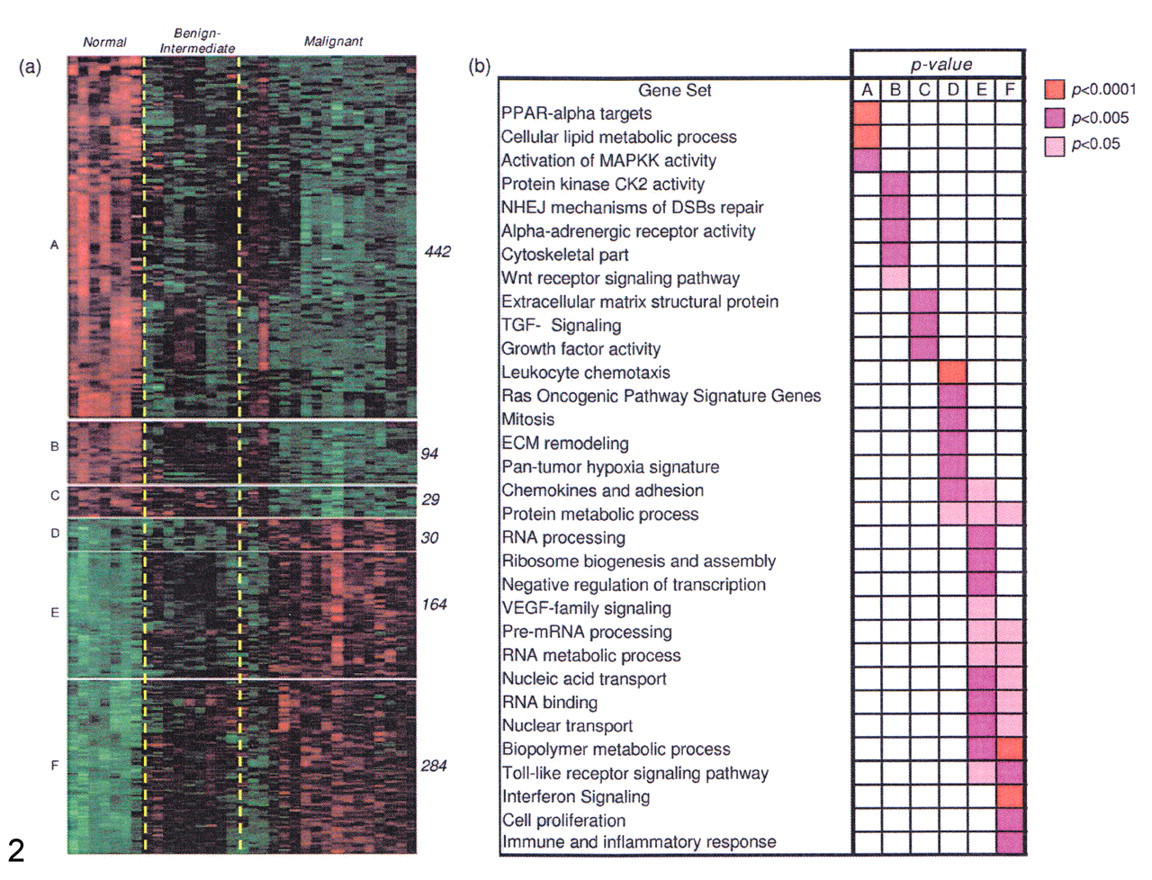
Identification of expressed gene classes in canine mammary samples analyzed with a canine-specific whole genome cDNA microarray. (a) Hierarchical clustering of dog samples based on a subset of 1,043 genes selected by one-way analysis of variance (q value < 0.001). For display purposes, samples in each class (normal, intermediate–benign, malignant) were clustered separately and arranged from normal (left) to malignant (right). Genes were categorized into 6 groups according to their pattern of expression (see description in the text) and hierarchically clustered separately. The gene size of each group is indicated to the right of each cluster. (b) Representation of the significance of gene set enrichment for the 6 gene groups. Enrichment scores are computed by one-sided Fisher exact test, using the entire set of orthologous genes represented on both human and dog microarray platforms as the reference population. This figure is reproduced with permission from BMC Genomics, 68 BioMed Central. All rights reserved. We would also like to thank Dr Uva for this illustration.
In the study by Uva et al, 68 a number of genes were differently expressed in more malignant subtypes. First, genes related to activation of the MAPK pathway, including the ELK3 transcription factor, were shown to be downregulated, as has been found in, for example, human malignant mesothelioma and cervical cancer cells, suggesting a potential role for these genes as tumor suppressors. 46,69 Second, genes involved in DNA repair, such as genes controlling protein kinase CK2 activity and the WNT receptor signaling pathway, were downregulated in malignant tumors. Third, genes upregulated in malignant phenotypes were found in angiogenic pathways (VGEF family) and the toll-like receptor (TLR) signaling pathway, known to promote the malignant transformation of normal epithelial cells in various tumors. Finally, genes responsible for the immune and inflammatory response and genes involved in interferon signaling were found to be overexpressed in the intermediate and malignant tumors. The authors admitted that their findings might have reflected the presence of inflammatory cells in these histologies, thus entailing expression from the stromal compartment instead of tumor tissue. However, it is possible that a significant proportion of these genes (CXCL16, IFI44, IFNAR1, IFNAR2, IFNGR2, ISG15, MX1, and STAT2) might also originate from tumor cells, as has been shown in humans. 66
Conclusion
The many human susceptibility gene studies in BC performed in the past decade are attracting company from an emerging interest for the same features in CMTs—owing to the access of a complete genome sequence, the high homology between the species, the relative “simplicity” of the canine genome concerning the possibility of using fewer SNPs to find candidate regions of the genome, the similar pattern of histologies, the similar biologic clinical behavior, and finally, the formation of spontaneous tumors in the dog.
It is said that the multifactorial nature of BC raises more hype among statistical geneticists than hope among clinical geneticists. The large number of anticipated susceptibility factors, their low predictive value, and the high frequency of these variants in the general population make these findings of limited use in clinical practice. 80 It is unlikely that new major-impact genes will be discovered in human BC; if this were the case, the high-resolution large GWAS recently performed would have indicated such discoveries. The structure of the canine genome (Fig. 1) may offer a greater chance of finding more major gene candidates responsible for cancer development, valid also for human oncology.
Even if new major susceptibility cancer genes are not found, the GWAS era should pick up several contributing genes to tumor development, which will give more information on oncogenesis and likely benefit drug development and early diagnostics. In the post-GWAS era in human medicine, massive parallel sequencing technology now allows the development of studies unachievable a few years ago. A major success in the use of this technology was the resequencing of the whole human genome where the results showed a higher complexity level of the human genome, with the appearance of many new variants, short-length insertions and deletions, and even small inversions. 79 This technique has already been used in human BC, where Rosa-Rosa et al reported 8 new candidates for functional studies and speculated that this new strategy could be a valid sequel to the GWAS analysis. 58 Still being expensive and not yet refined in humans, massive parallel sequencing will likely be the next step to increase the resolution in genome analysis in dogs. Accompanied by the specific multigene marker expression analyses now performed in both species, the new information should in turn lead to the ultimate goal of improving treatment outcomes and learning more about how known environmental risk factors interact with the polygenetic causes of mammary tumors in both human and dog.
Footnotes
The authors declared that they had no conflicts of interest with respect to their authorship or the publication of this article.
Mammary cancer comparative research was funded by the European Commission’s Seventh Framework Programme for Research and Technological Development (LUPA grant No. GA-201370) and the AGRIA Pet Insurance Research Foundation, Linnea and Axel Ericsson’s Foundation, and the Amanda Personnes Research Foundation, all 3 from Sweden.