
Abstract
Telomerase deficiency induces early senescence and defects in proliferating cell populations, but in mice it has not been associated with inflammatory bowel disease. Genetically engineered mice lacking either telomerase reverse transcriptase (TERT) or telomerase RNA were examined for chronic diarrhea and wasting. Affected mice had pasty stools, thickened nondistensible colon walls, and contracted ceca. Histologically, the cecal mucosa was largely replaced by inflammatory infiltrate consisting of plasma cells, neutrophils, lymphocytes, and macrophages with marked widespread fibrosis and ulceration. Remaining epithelium was disorganized and hyperplastic, with multifocal dysplasia. Colonic mucosa was markedly hyperplastic with similar inflammation and epithelial dysplasia. Multifocal adenomatous hyperplasia, but no inflammation, was present in the small intestine. Microaerophilic spiral bacteria with 16S rRNA gene sequences identical to Helicobacter mastomyrinus were isolated from the colon and cecum. Severe granulomatous typhlocolitis without epithelial dysplasia developed in germ-free recombination-activating gene (RAG) knockout (KO) recipients of CD4+ T cells and inoculated with cecal contents from affected TERT KO mice and in specific pathogen-free recipient RAG KO mice and interleukin-10 KO mice inoculated with H mastomyrinus. Typhlocolitis in mice given H mastomyrinus was more severe than in mice given Helicobacter hepaticus. Telomerase-deficient mice are susceptible to helicobacter-associated typhlocolitis. H mastomyrinus causes severe disease in susceptible mouse strains.
Murine enterohepatic helicobacters are now generally recognized as opportunistic pathogens that cause hepatitis, typhlocolitis, and hepatic and colonic neoplasia in susceptible mouse strains. 16,34,52 There remains considerable controversy regarding the role of these organisms as primary pathogens, however, and the relative pathogenicity of different helicobacter species is not well studied. Of the 11 species of enterohepatic helicobacter species thus far identified in rodents, Helicobacter hepaticus is the most commonly isolated and best studied. It was first identified in association with hepatitis and hepatic carcinoma in A/J mice, 49,52 and it has been shown to be associated with typhlocolitis in a number of mouse strains that have altered immune function. 8,15,17,28,30,50 Other species associated with inflammation of the lower bowel in mice include Helicobacter typhlonicus, 10,19 Helicobacter bilis, 43 Helicobacter muridarum, 24 Helicobacter rodentium, 10,42 Helicobacter trogontum, 53 and Helicobacter ganmani, 57 but there are few comparative studies on the pathogenicity of different helicobacter species. In addition, some studies have described inflammatory bowel disease (IBD) in mice in the absence of detectable helicobacters, 12,20 and others have implicated nonhelicobacter species in lower bowel inflammation. 2,39 Thus, there remains considerable debate regarding whether or not these organisms are primary pathogens, and therefore they are not routinely eliminated in some mouse colonies. In fact, a recent survey revealed the presence of one or more helicobacter species in 88% of 34 institutions worldwide, 46 suggesting that concern regarding elimination of these organisms remains low.
In addition to their role in naturally occurring disease in infected colonies, lower bowel helicobacters have attracted interest as models of IBD in humans. The best studied of these models are interleukin-10 (IL-10) knockout (KO) mice 7,28,53 and severe combined immunodeficient (SCID) or recombination-activating gene (RAG) KO mice that are adoptively transferred with CD4+ effector T cells from congenic mice (recipient RAG KO or SCID mice). 24,27 Both these models lack regulatory T-cell function, and both develop severe typhlocolitis in response to H hepaticus and several other enteric murine helicobacters. Study of these as well as several other models involving mutant mouse strains and lower bowel helicobacters has indicated that a common factor in susceptibility to helicobacter-associated IBD in mice is defective immunoregulation and that severe inflammation may in some cases lead to neoplastic transformation (for reviews, see Strober et al 45 and Wirtz and Neurath 54 ). Thus, these models are of interest in the investigation of the role of immune dysfunction and bacterial colonization in IBD and colon cancer in humans.
In this report we describe a spontaneous outbreak of severe inflammatory lower bowel disease in telomerase-deficient mice. A single helicobacter species, Helicobacter mastomyrinus, was identified in affected mice, and both IL-10 KO mice and recipient RAG KO mice developed severe typhlocolitis when inoculated with H mastomyrinus from telomerase-deficient mice.
Methods
Mice
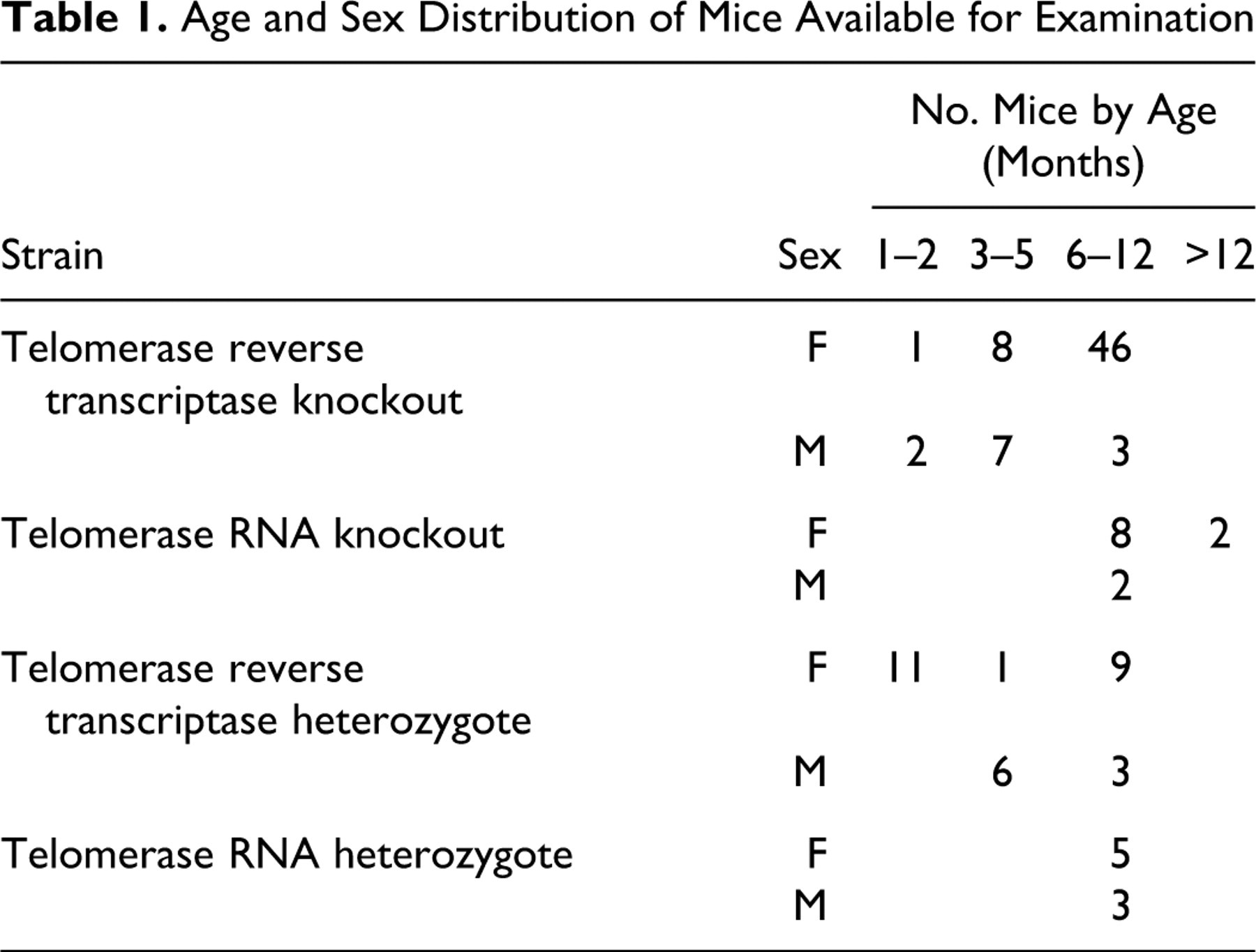
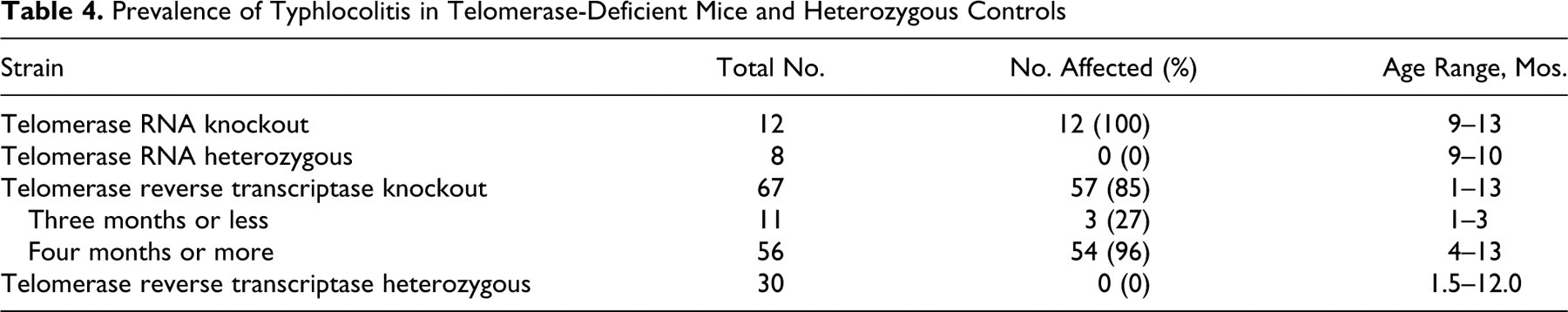
Telomerase-deficient mice were from the breeding colony at the University of Michigan and were kindly supplied by Dr Sem Phan. The colony was established in 2003, and sentinels were screened quarterly for mouse pathogens by the University of Michigan Unit for Laboratory Animal Medicine (for a list of pathogens, see http://ulam.med.umich.edu/services/healthcare/surveillance.html). Note that the University of Michigan does not routinely screen for helicobacter. Two mutant mouse strains were examined. Telomerase reverse transcriptase (TERT) KO mice were engineered to contain a partial deletion of the TERT gene. 31,56 These mice expressed telomerase RNA (TR) but not telomerase enzyme. mTR KO mice 5 lacked the TR component but carried wild-type TERT. Both mouse strains were functionally telomerase deficient, 5,56 were on a C57BL/6 background, and were routinely maintained by breeding of heterozygotes. On some occasions, homozygotes were bred, but offspring of homozygote matings were not used as breeders. Thus, in all cases, mice were offspring of heterozygote matings or first-generation offspring of homozygote matings. The mice were maintained in microisolator cages and fed standard rodent chow and water ad libitum. No experimental procedures were performed on any of the mice. Sick mice were submitted to the pathology service of the Unit for Laboratory Animal Medicine for necropsy. Table 1 indicates the genotype, sex, and age distribution of the mice available for examination.
Age and Sex Distribution of Mice Available for Examination
Male and female germ-free RAG-1 KO mice on a C57BL/6 background were from the University of Michigan Germ Free Life Laboratory. They were maintained in sterile bubble isolators, and they remained bacteriologically sterile until removed from the isolators and inoculated with cecal contents. Germ-free mice were conventionalized by oral inoculation with feces from normal helicobacter-free C57BL/6 mice (Jackson Laboratories, Bar Harbor, ME) and removal to microisolator housing. IL-10 KO and congenic C57BL/6 mice were from the University of Michigan breeding colony, housed separately from the telomerase-deficient mice, and were helicobacter-free as determined by polymerase chain reaction (PCR) screening. All mice were on a C57BL/6 background. All animal experiments were approved by the University of Michigan Committee on Use and Care of Animals.
Bacterial Culture and Inoculation
For primary isolation of helicobacter from feces, colon, or cecum, fresh samples were homogenized in 500 μl of trypticase soy broth (TSB), passed through a 0.45-μm syringe filter, collected in a sterile tube, and streaked onto TSA-CVA plates (trypticase soy agar, 5% sheep blood, 20 μg/ml cefoperazone, 10 μg/ml vancomycin, 2 μg/ml amphotericin B). The culture was incubated at 37°C under microaerobic conditions for 10 days, and cultures were identified as H mastomyrinus by culture morphology, microscopic examination, and cloning and sequencing of the 16S rRNA gene. H mastomyrinus strains AY742307 and AY631955, kindly supplied by Dr Jim Fox, were used for comparisons. Microaerobic conditions were generated as previously described 36 by evacuation of vented GasPak jars without a catalyst after evacuation to 20 mm Hg and equilibration with a gas mixture consisting of 80% N2, 10% CO2, and 10% H2.
H mastomyrinus isolated from TERT KO mice and H hepaticus strain 3B1(ATCC 51449) were cultured as described above on trypticase soy agar plates containing 5% sheep blood without antibiotic supplement. For mouse inoculation, cultures were incubated as above for 4 days, and bacteria were scraped from plates and resuspended in TSB to an optical density of 1.0 at 600 nm (approximately 108 colony-forming units per ml). Mice were orally inoculated with 0.15 ml of the bacterial suspension or with sterile TSB. Bacteria recovered from feces or cecal contents of infected mice were identified as H mastomyrinus or H hepaticus by culture morphology, microscopic examination, PCR, restriction fragment length polymorphism (RFLP) analysis, and sequencing (H mastomyrinus only).
DNA Isolation and PCR
To identify bacteria in cecum, colon, or feces, genomic DNA was extracted with the DNeasy Blood & Tissue Kit (Qiagen, Valencia, CA) using a modified protocol. Modifications included (1) adding a bead-beating step using UltraClean fecal DNA bead tubes (Mo Bio Laboratories, Inc, Carlsbad, CA) shaken with a Mini-Beadbeater-8 (BioSpec Products, Inc, Bartlesville, OK) at the “homogenize” setting for 1 minute; (2) increasing the amount of buffer ATL used in the initial steps of the protocol (from 180 μl to 360 μl); (3) increasing the volume of proteinase K used (from 20 μl to 40 μl); and (4) decreasing the amount of buffer AE used to elute the DNA at the end of the protocol (from 200 μl to 100 μl). 1 DNA was amplified with conserved 16S rRNA helicobacter genus-specific primers (C05: 5′ ACT TCA CCC CAG TCG CTG 3′ and C97: 5′ GCT ATG ACG GGT ATC C 3′). 42 The cloning reaction was performed as described previously 27 with illustra PuReTaq Ready-to-Go PCR Beads (GE Healthcare, Piscataway, NJ). Each 25-μl PCR mixture contained 20 pmol of each primer, 200 μM of each deoxynucleoside triphosphate, and 1.5 U of Taq DNA polymerase in a final concentration of 10 mM Tris-HCl, 50 mM KCl, and 1.5 mM MgCl2. To identify cultured bacteria, DNA was extracted with the Easy-DNA Kit (Invitrogen, Carlsbad, CA) per the manufacturer’s instructions and amplified with helicobacter genus-specific primers C05 and C97 (above) and with species-specific primers (C60: 5′ AGA ACT GCA TTT GAA ACT ATG AG 3′ and C61: 5′ CAG TAT TGC GTC TCT TTG TA 3′). 41
Bacteria in situ were identified by cloning and sequencing of PCR products. For this, PCR amplicons were purified with the illustra MicroSpin S-400 HR Columns (GE Healthcare) as directed by the manufacturer. Purified products were ligated into the TOPO 4 vector (K4575-01, Invitrogen) according to manufacturer’s specifications and transformed into Escherichia coli. Individual colonies were picked following overnight incubation on Luria-Bertani (LB) plates with carbenicillin (50 μg/ml), then cultured overnight in LB broth freeze media with carbenicillin (50 μg/ml). Vector specific primers (M13F: 5′-CAGTCACGACGTTGTAAAACGACGGC-3′ and M13R: 5′-CAGGAAACAGCTATGACCATG-3′) were used to screen these colonies for bacterial clones containing the appropriate 1.2-kb amplicon insert. Clones were sequenced with M13F and M13R primers at the University of Michigan’s Sequencing Core.
Bacterial cultures were identified by RFLP fingerprinting and sequencing of PCR amplicons. For RFLP fingerprinting, amplicons from the genus-specific primers (1.2 kb) were digested with HhaI. H hepaticus yielded 3 bands of 150, 250, and 800 base pairs. H mastomyrinus yielded 3 bands of 300, 400, and 600 base pairs, as described by Shen et al. 41 For sequencing, species-specific amplicons were submitted to the Sequencing Core at the University of Michigan. The sequences were assessed for quality, trimmed, aligned, and classified using the online tools of the Ribosomal Database Project (http://rdp.cme.msu.edu). 11,48
Immunochemistry
Identity of inflammatory cells in formalin-fixed, paraffin-embedded sections was determined by morphology in hematoxylin and eosin–stained sections and by immunoreactivity to CD3 (T cells), CD45R/B220 (B cells), F4/80 (macrophages), or IRF-4 (plasma cells). 52 Immunostaining was performed on an automated immunostainer (intelliPATH, Biocare, Concord, CA). Antigen retrieval for macrophage detection was performed by 10 minutes of trypsin digestion (Biocare) at room temperature. Retrieval for T cells, B cells, and plasma cells was performed by heat-induced retrieval at 95°C in a decloaking chamber (Biocare) using commercial solutions (T cells, Diva, Biocare; B cells, Rodent Decloaker, Biocare; plasma cells, citrate buffer, Biogenex, San Ramon, CA). T cells were labeled with rabbit monoclonal anti-CD3 (RM9107-S, Thermo-Scientific, Waltham, MA) at 1:500 with 1 hour of incubation. B cells were labeled with rat monoclonal anti-CD45R/B220 (550286, BD Biosciences, San Jose, CA) at 1:200 for 30 minutes of incubation. Macrophages were labeled with rat monoclonal anti-F4/80 (ab6640, Abcam, Cambridge, MA) at 1:100 with 1 hour of incubation. Plasma cells were labeled with goat polyclonal anti-IRF-4 (sc-6059, Santa Cruz Biotechnology) at 1:500 with 1 hour of incubation. Immunodetection was by a horseradish peroxidase–conjugated polymer system with diaminobenzidine for rat and rabbit primary antibodies (Biocare Promark series: Rat-on-Rodent, Rabbit-on-Rodent) and an alkaline phosphatase–conjugated polymer system (Biocare Goat Polymer Detection) with Biocare Vulcan fast red for goat antibodies. Negative controls were naïve serum from the species of the primary antibody. Sections were counterstained with hematoxylin.
Transmission Experiments
For determination of bacterial pathogenicity, 3 groups of mice were used: germ-free RAG KO mice removed from the sterile germ-free isolators and inoculated with cecal contents from TERT KO mice; germ-free RAG KO that had been conventionalized by removal from the germ-free isolators and housed in specific pathogen-free conditions for 30 days before inoculation with H mastomyrinus isolated from telomerase-deficient mice or with H hepaticus strain 3B1; and IL-10 KO mice inoculated with H mastomyrinus or H hepaticus. Two weeks following bacterial inoculation, RAG KO mice were intraperitoneally inoculated with 106 CD4+ enriched splenocytes from C56BL/6 mice. CD4+ cell enrichment was performed by magnetic bead separation as previously described. 13 Cells isolated in this way contain > 95% CD4+ cells and B cells, and CD8+ T cells are below the limit of detection. Control mice were either uninfected or untransferred. Table 2 indicates mouse groups and the number of mice per group.
Mouse Groups Experimentally Inoculated With Helicobacter

Tissue Collection
Telomerase-deficient KO and heterozygous mice were euthanized when severe diarrhea was noted or as age-matched controls. Experimentally infected mice were euthanized at intervals indicated in Table 2. At necropsy of telomerase-deficient and heterozygous control mice, gross lesions were recorded, and sections of all levels of intestinal tract, as well as kidney, liver, lung, heart, and spleen, were immersion fixed in 10% neutral buffered formalin and embedded in paraffin. Five-micron sections were stained with hematoxylin and eosin or Warthin–Starry silver stain and examined for the presence or absence of morphologic lesions or bacteria in the intestinal lumen. Cecal contents and feces were collected for culture and PCR identification of helicobacter.
At necropsy of experimentally infected mice, stomach, small intestine, cecum, and colon were immersion fixed in 10% neutral buffered formalin and embedded in paraffin. Cecum was emptied of contents and embedded in its entirety with the ileocecal junction toward the surface of the block. Sections of proximal, middle, and distal colon were embedded and cut in longitudinal sections. Five-micron sections were stained with hematoxylin and eosin or Warthin–Starry silver stain, and lesions were semiquantified as described below.
Histologic Scoring
Sections of colon and cecum from experimentally infected mice were scored for severity of lower bowel inflammation, as indicated in Table 3 . All sections were coded and scored blind. Lesions scored were as follows: mucosal hypertrophy and hyperplasia, neutrophilic infiltrate, mononuclear inflammatory infiltrate (lymphocytes, plasma cells, or histocytes), and adenitis, defined as dilation of glands with cellular debris and neutrophils. Severity was estimated on a scale of 0 to 3, as shown in the table. In addition, the presence or absence of serositis (indication of transmural inflammation) was recorded and scored as absent (0) or present (1), and organized mucosal lymphoid follicles (mucosa-associated lymphoid tissue) were scored as absent (0), present (1), or hyperplastic (2). The total score for each section was determined by adding the scores for the individual lesions.
Histologic Scoring of Cecum and Colon of Experimentally Infected Mice
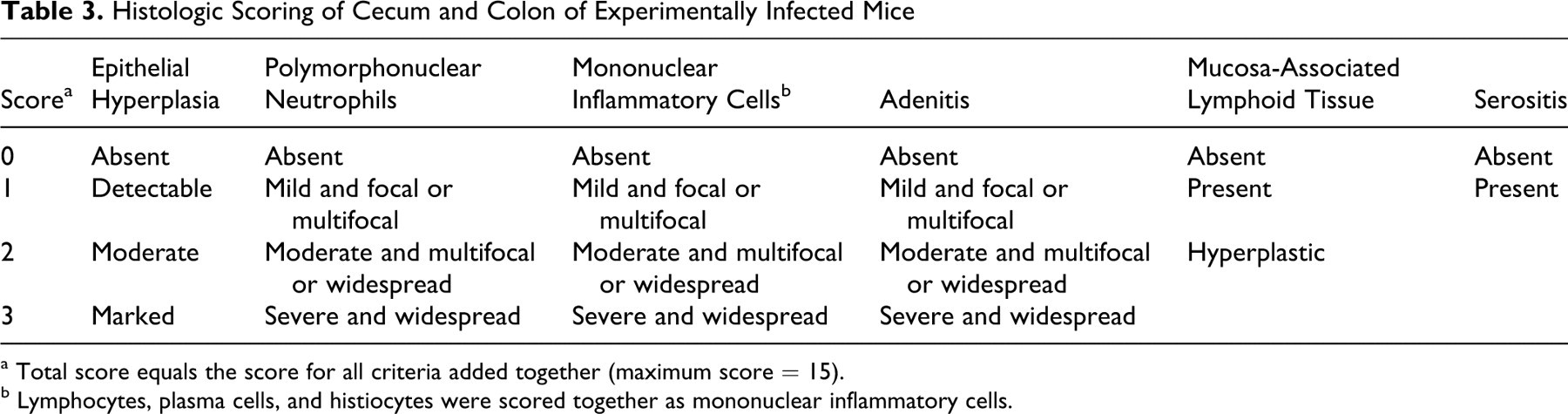
a Total score equals the score for all criteria added together (maximum score = 15).
b Lymphocytes, plasma cells, and histiocytes were scored together as mononuclear inflammatory cells.
Statistics
Groups were compared by Mann–Whitney U test (2 groups) or by analysis of variance with Bonferroni corrections for multiple groups. Error bars in charts indicate standard error of the mean.
Results
Wasting and Typhlocolitis in Telomerase-Deficient Mice
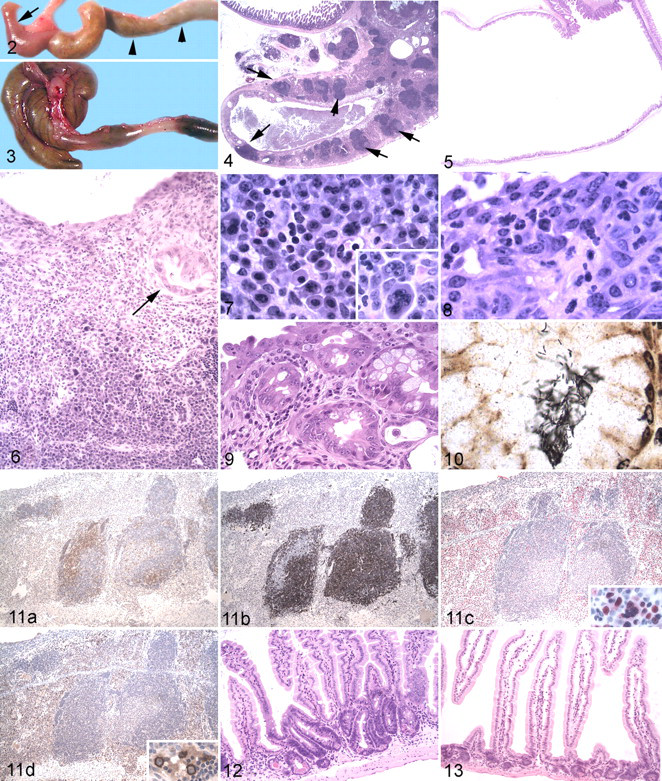
Affected mice appeared thin with little body fat and wasted musculature. Mean body weight in female TERT KO mice, the only group that contained enough control mice for comparison, was significantly less than that of age-matched heterozygous controls (Fig. 1). Grossly, ceca in affected mice were small and firm with thickened walls and minimal to absent lumens (Fig. 2). In contrast to unaffected heterozygous controls (Fig. 3), colons of affected mice were distended with soft, pasty stools and had thickened walls. Gross lesions were confined to the large intestine, except that one mouse had unilateral hydronephrosis, considered to be a background lesion in C57BL/6 mice. Heterozygous controls had no gross lesions.
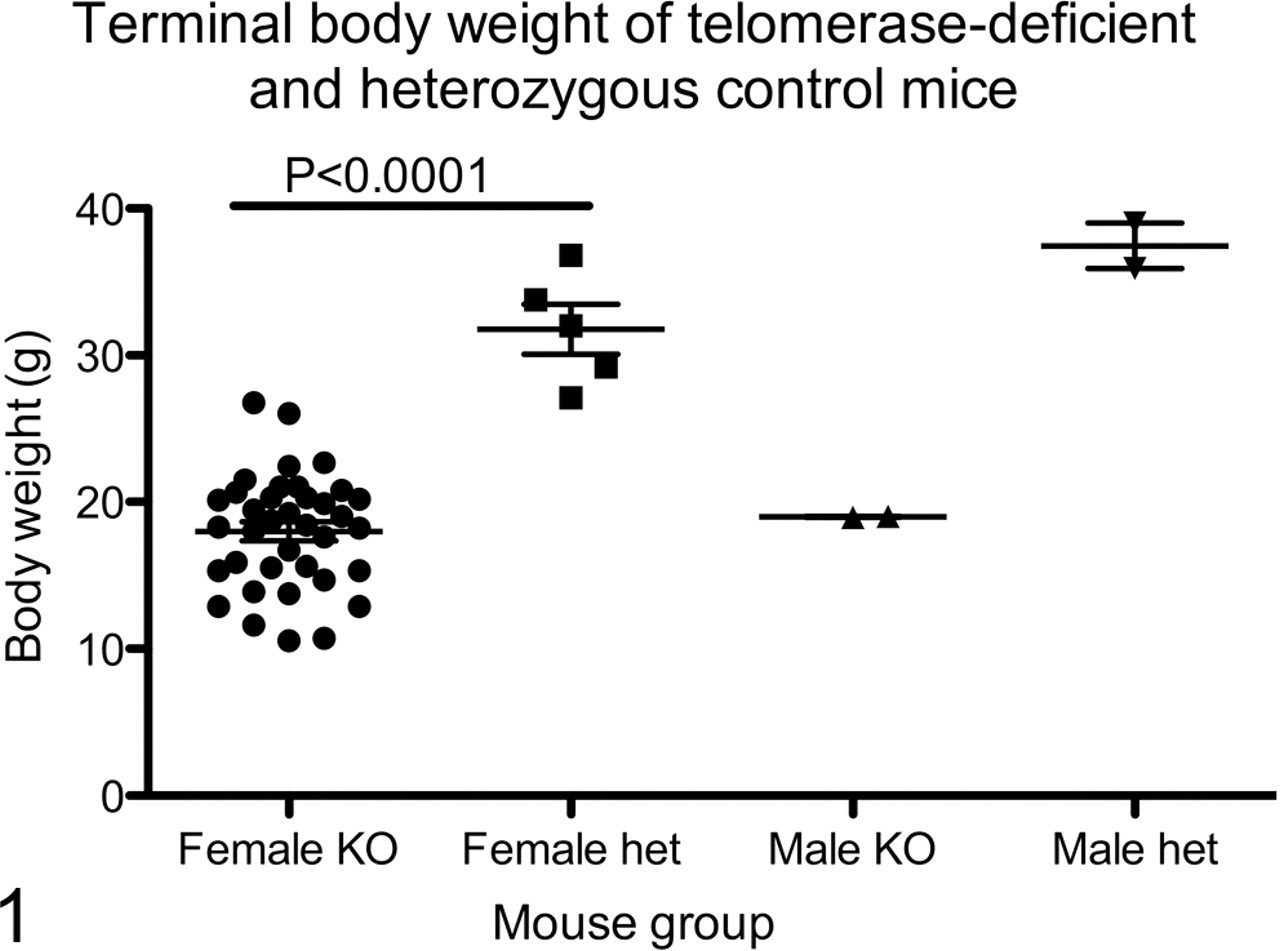
Body weights of telomerase reverse transcriptase knockout (KO) and heterozygous (het) control mice. Mice were weighed immediately following euthanasia. Each symbol represents one mouse.
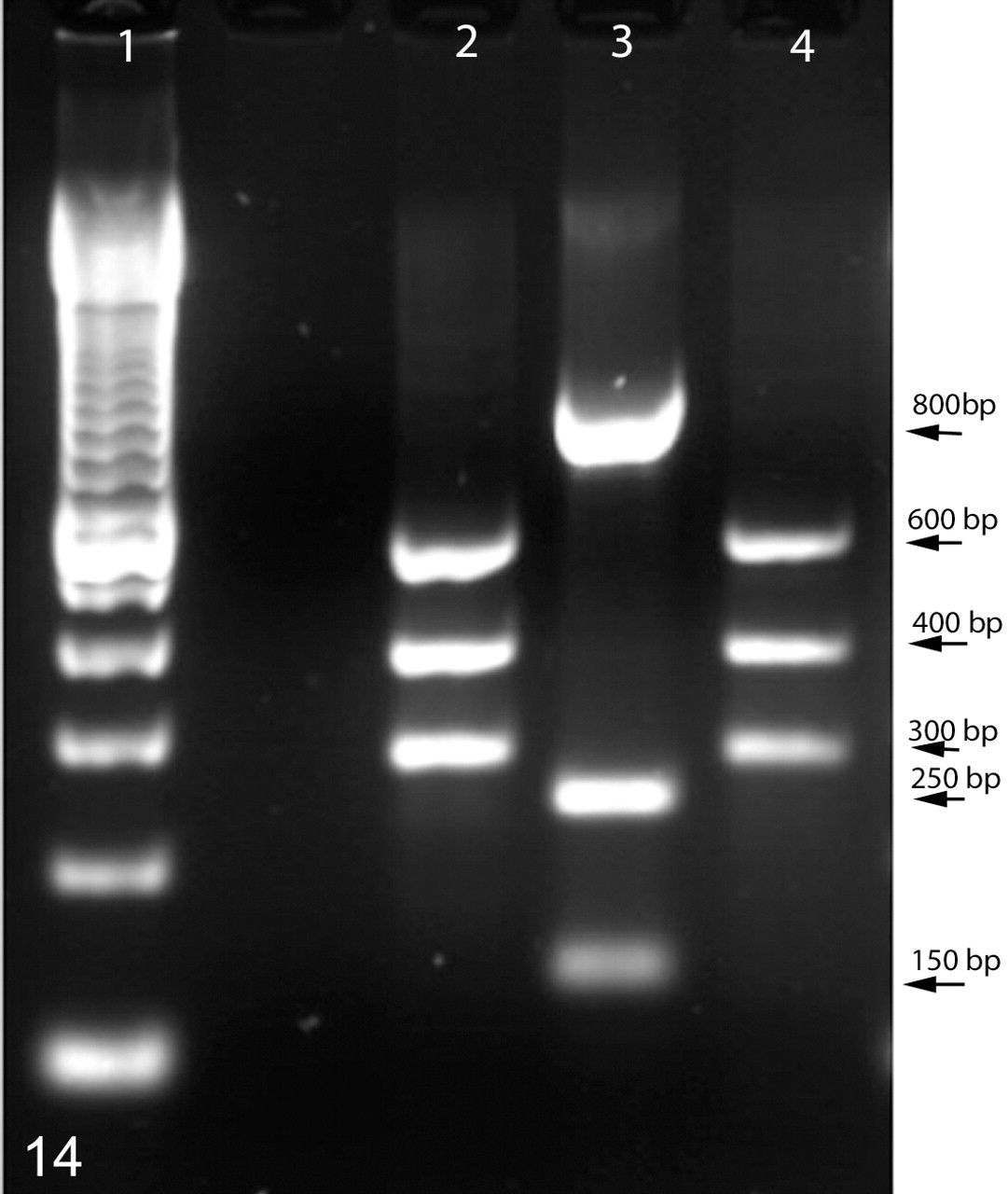
Restriction fragment length polymorphism of helicobacter strains. The rRNA gene was polymerase chain reaction amplified and digested with HhaI, and fragments were separated on an agarose gel. Lane 1, size markers; lane 2, Helicobacter mastomyrinus isolated from a telomerase reverse transcriptase knockout mouse; lane 3, Helicobacter hepaticus 3B1; lane 4, cloned H mastomyrinus rRNA gene from a telomerase reverse transcriptase knockout mouse. Approximate band sizes are indicated to the right of the figure; bp, base pairs.
Histologically, the cecal walls were markedly thickened, and the epithelium was largely replaced by inflammatory infiltrate, mucosal lymphoid follicles, and fibrous connective tissue (Fig. 4). The cecal lumens were markedly reduced in size compared to those of control mice (Fig. 5). Typhlitis was characterized by severe transmural inflammation (Fig. 6) consisting primarily of numerous activated plasma cells that were occasionally binucleate or multinucleate (Fig. 7), granulocytes (mostly neutrophils with a few eosinophils; Fig. 8), and fewer macrophages and lymphocytes. Similar infiltrate was present, extending into the surrounding serosa and within local lymph nodes. In some mice there was granulocytic hyperplasia of splenic hepatopoietic tissue.
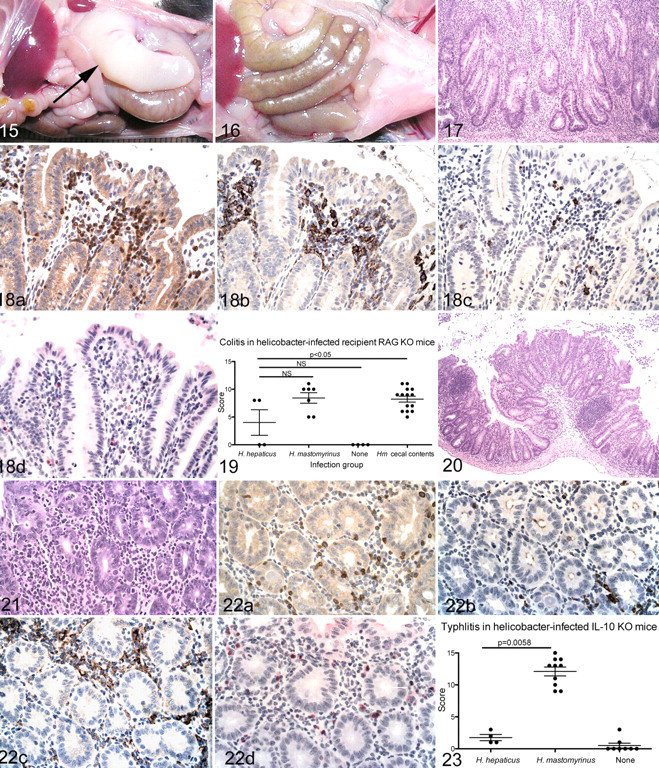
The Cecal epithelium was largely absent and replaced by fibrosis and inflammation, with only a few retained proliferative epithelial nodules (Fig. 6). In addition to ulceration and loss of glands, epithelial changes included widespread proliferation characterized by piling up of nuclei and many mitotic figures. Focal epithelial dysplasia was present in some proliferating glands and was characterized by loss of cell polarity, anisocytosis, anisokaryosis, and cellular atypia (Fig. 9). Remnant proliferating glands were often dilated with necrotic cellular debris. In the colons, there was marked epithelial hyperplasia and chronic neutrophilic inflammation, but epithelial dysplasia and ulceration were less prominent than in the ceca. Warthin–Starry stains of cecum and colon revealed dense populations of bacteria in the lumen and glands of affected mice. Bacteria were of mixed morphologic types, but most bacteria in the glands were elongated narrow spirals, morphologically compatible with H mastomyrinus as previously described 41 (Fig. 10). Similar organisms were present in unaffected heterozygous mice but were not associated with inflammation.
Most infiltrating inflammatory cells were identified as plasma cells based on morphology and immunoreactivity to IRF-4 but not B220 or F4/80 (Fig. 11). Immunochemical staining revealed that the remaining mononuclear inflammatory cells were B cells (mostly confined to follicles) with fewer T cells and scattered macrophages (Fig. 11). In the small intestine, inflammation was absent, but foci of crypt epithelial proliferation were present throughout the duodenum, jejunum, and ileum in telomerase-deficient mice (Fig. 12) but not heterozygous mice (Fig. 13). These were characterized by increased cell density and increased basophilia with scattered mitotic figures. There were no histologic lesions in stomach, heart, lung, kidney, or liver.
Table 4 indicates the number of mice affected in each group. Of the 79 telomerase-deficient mice examined, all but 10 were affected (87%). Interestingly, of the 10 unaffected mice, 8 were 3 months old or younger, suggesting that older mice were more susceptible to disease or that disease was progressive (ie, becoming worse with age), but this could not be statistically confirmed. None of the heterozygous mice had gross or histologic evidence of typhlocolitis, although helicobacter-like bacteria were present in Warthin–Starry-stained sections of cecum and colon (not shown).
Prevalence of Typhlocolitis in Telomerase-Deficient Mice and Heterozygous Controls
Isolation and Identification of H mastomyrinus
A single species of helicobacter was isolated from affected telomerase-deficient mice and identified as H mastomyrinus on the basis RFLP and sequencing of the 16S rRNA gene. RFLP of genus-specific PCR amplicons from recovered bacteria revealed 3 bands identical in size to those described for H mastomyrinus by Shen et al 42 and distinct in size from those of H hepaticus (Fig. 14). Sequencing of helicobacter-specific rRNA clones and PCR amplicons from helicobacter recovered from affected mice revealed a single species of helicobacter in all mice tested. Sequence comparison revealed > 99% sequence identity with each other and with the H mastomyrinus–type strains, AY742307 and AY631955, leading to their identification as H mastomyrinus.
Recipient RAG KO Mice
RAG KO mice were inoculated with cecal contents from affected TERT KO mice, pure cultures of H mastomyrinus from TERT KO mice, or pure cultures of H hepaticus and given CD4+ T cells by adoptive transfer. All recipient mice inoculated with either H mastomyrinus or cecal contents developed severe typhlocolitis that was clinically similar to disease in naturally infected TERT KO mice. Mice developed soft stools, and 1 mouse became moribund and was euthanized 3 weeks after adoptive transfer. All the remaining mice survived for the 8-week duration of the experiment. Grossly, mice had dilated ceca and colons with thickened walls (Fig. 15) similar to those of affected TERT KO mice except that the ceca were somewhat less atrophied in the recipient RAG KO mice compared to the naturally infected TERT KO mice. Uninfected recipient RAG KO mice had no gross lesions (Fig. 16). Histologic lesions differed somewhat between recipient RAG KO mice and naturally infected telomerase-deficient mice. In recipient RAG KO mice (like telomerase-deficient mice), typhlocolitis was characterized by marked, widespread infiltration with granulocytes and mononuclear cells and marked epithelial proliferation (Fig. 17). In recipient RAG KO mice, however, mucosa-associated lymphoid follicles were absent, presumably because insufficient numbers of B cells were present in the transferred cell population (see Methods). Epithelial dysplasia was absent in infected recipient RAG KO mice (Fig. 17).
The distribution of inflammatory cell types in infected recipient RAG KO mice also differed from that of naturally infected telomerase-deficient mice. In recipient RAG KO mice, immunochemical identification of cell subsets revealed that most infiltrating mononuclear inflammatory cells were immunoreactive for either CD3 or F4/80 and negative for IRF-4, indicating that most were T cells or macrophages (Fig. 18) rather than plasma cells, as in telomerase-deficient mice. Multinucleated cells in these mice were of macrophage origin, as indicated by immunoreactivity for F4/80 but not IRF-4. As indicated in Figure 18, only rare cells were immunoreactive for B220 or IRF-4. Because the cell population transferred to these mice was highly enriched in T cells 14 and because recipient mice do not develop either B-cell follicles or serum immunoglobulin G antibody, 14 these cells were presumed to be either small numbers of residual cells in the transferred population or immature cells that cross-reacted with the B-cell and plasma-cell markers.
Like H mastomyrinus–infected mice, recipient RAG KO mice infected with H hepaticus developed typhlocolitis, but it was less severe than that in mice infected with H mastomyrinus. This interpretation is based on the following observations: Gross lesions were not apparent in mice given H hepaticus; only 2 of 4 mice developed colitis; mean colitis scores were significantly less than those in mice given TERT cecal contents; and of the 2 mice with lesions, scores were less than those in mice given either H mastomyrinus or TERT KO cecal contents (Fig. 19).
IL-10 KO Mice
IL-10 KO mice inoculated with H mastomyrinus cultured from TERT KO mice developed severe typhlocolitis within 2 weeks of inoculation. Lesions were similar to those in naturally infected TERT KO mice. Grossly, mice had soft stools and thickened, dilated colons and ceca. Inflammation was transmural, accompanied by epithelial proliferation and mucosal lymphoid follicles (Fig. 20), and characterized by lymphocytes, macrophages, and neutrophils (Fig. 21). As in the RAG KO mice, most infiltrating cells were identified by immunochemistry as macrophages, with fewer T cells, B cells, and plasma cells (Fig. 22). Multinucleated giant cells were rarely present. As in TERT KO mice, mucosa-associated lymphoid nodules were prominent, but as in recipient RAG KO mice, focal epithelial dysplasia was absent. Small intestine was normal in IL-10 KO mice.
In contrast to mice inoculated with H mastomyrinus, H hepaticus–infected IL-10 KO mice developed minimal to mild typhlocolitis, even by 27 days after inoculation. Typhlitis scores in H mastomyrinus–infected mice were significantly greater than scores in H hepaticus–infected mice, which were not significantly different from those of uninfected mice (Fig. 23). H hepaticus– and H mastomyrinus–infected C57BL/6 control mice had no gross or histologic lesions.
Discussion
The major findings of this study are as follows: (1) Telomerase-deficient mice are susceptible to helicobacter-associated typhlocolitis; (2) H mastomyrinus is potentially pathogenic in susceptible mouse strains; and (3) H mastomyrinus is more pathogenic than H hepaticus in 2 models of IBD.
Lower bowel helicobacters are a common contaminant of laboratory mouse colonies. They do not cause disease in most inbred strains but have been associated with inflammatory bowel lesions in a number of strains of mutant mice. H hepaticus, the most commonly identified murine helicobacter and the best studied, was first identified in association with hepatitis and hepatocellular carcinoma in male A/J mice 49,52 and subsequently shown to be associated with IBD in several mutant mouse strains. 8,9,15,17,28,30,50 Subsequently, several other species of lower bowel helicobacters have been associated with IBD in susceptible mouse strains. 10,19,24,42,43,53,57 Their direct role in disease remains controversial, however. In some studies of IBD in mice, helicobacter species were not identified, 12,20 and bacterial genera other than helicobacter have been associated with bowel inflammation in mice. 2 Some authors have suggested that murine lower bowel helicobacters may cause disease indirectly by inducing damaging host responses to co-colonizing enteric organisms. 23 Thus, it is likely that factors in addition to helicobacter contribute to disease and that helicobacter species are one of several members of the indigenous murine microbiota that cause or exacerbate inflammatory lesions in susceptible mouse strains.
H mastomyrinus was originally isolated from a multimammate rat (Mastomys natalensis) and a colony of laboratory mice. 41 It has subsequently been identified in several mouse colonies, 6,46 but its pathogenicity in helicobacter-susceptible mouse strains was not evaluated. In the current study, we demonstrated that H mastomyrinus isolated from mice with typhlocolitis induced lower bowel inflammation in 2 commonly studied models of helicobacter-associated IBD, thus demonstrating its pathogenic potential. Furthermore, in recipient RAG KO mice, lesions due to H mastomyrinus were significantly more severe than those due to H hepaticus, suggesting that helicobacter species may differ in their pathogenic potential and that H mastomyrinus is more pathogenic that H hepaticus.
Differences in pathogenicity of different helicobacter species have been described. One study found small differences in pathogenicity of H hepaticus and H bilis in 2 mouse models. 7 In another study, Helicobacter typhlonius was found to be more pathogenic than H rodentium in IL-10 KO mice, and the combination of 2 strains more pathogenic still. 10 In recipient RAG KO mice, H hepaticus was shown to be more pathogenic than Helicobacter ganmanii. 57 Conversely, in a different model, H hepaticus was associated with amelioration of disease, whereas H bilis was associated with exacerbation. 32 Thus, the reported outcome of infection by lower bowel helicobacters varies markedly among different studies. It is likely that pathogenicity varies according to the helicobacter species and to the characteristics of the mouse model used and that environment, host, and bacterial factors all contribute to the extent of disease.
In this study, H mastomyrinus caused severe typhlocolitis in all 3 mouse strains. Differences in infiltrating cell types is likely attributable to the specific immunologic defect of the individual models. Recipient RAG KO mice lack functional B cells 15 and regulatory T cells 37 compatible with the absence of lymphoid follicles and the large number of T cells and macrophages in the inflammatory infiltrate. IL-10 KO mice are defective in immunoregulation, likely accounting for T cells and macrophages in the infiltrate, but they do have functional B cells and develop mucosal lymphoid follicles, as do the telomerase-deficient mice. The specific immunologic defects in telomerase-deficient mice is not yet known, but the large number of plasma cells and B-cell follicles may implicate T-cell or regulatory-cell dysfunction.
The current study is, to our knowledge, the first to describe helicobacter-associated typhlocolitis in telomerase-deficient mice, and the pathogenesis of disease in this mouse strain is not yet known. Comparison with similar mouse models of helicobacter-associated IBD strongly implicates immune dysfunction, epithelial dysfunction, or both as common pathways to lower bowel inflammation. As noted above, the IL-10 KO and recipient RAG KO models are the best studied of the mouse models of helicobacter-associated IBD. However, many other mouse strains are susceptible to inflammatory disease of the lower bowel, and a number of models have been used to examine various aspects of IBD pathogenesis (for reviews, see Strober et al 45 ). These models markedly differ in mouse background strain, mutation, and experimental manipulation, but there are some common factors—notably, the functional defect responsible for inflammation is either failure of immunoregulation, damage to the epithelial barrier function, or both. In the models used in the current study, disease has been attributed to dysfunction of regulatory T cells (IL-10 KO) 45 or their absence (recipient RAG KO), 44 leading to a dysregulated hyperinflammatory response to a member of the enteric microbiota. Similarly, mice that are deficient in IL-2, IL-2 receptor, or transforming growth factor β are associated with failure of immunoregulation and IBD, whereas mice overexpressing tumor necrosis factor α and T-bet develop inflammation presumably due to a hyperinflammatory state. In a few models, such as the multidrug-resistant mice and glutathione peroxidase–deficient mice, disease has been attributed to leakiness of the intestinal epithelial barrier, presumably allowing contact of proinflammatory luminal contents with immune cells in the lamina propria. 45
Either immune or epithelial defects could contribute to IBD in telomerase-deficient mice. Several studies have described immunologic abnormalities that could lead to immune dysfunction in association with lack of telomerase. In humans and mice, telomerase deficiency leads to abnormal development of bone marrow stem cells and can lead to defects in hemopoietic cells. 4,25 Telomerase-deficient mice have fewer T cells and B cells than do C57BL/6 controls, and their lymphocytes are less responsive to stimulation in vitro, 22,29 suggesting that immune dysfunction is one outcome of telomerase loss. Aging studies have indirectly implicated telomerase in immunodysfunction and chronic inflammation. Telomere shortening with increasing age has been observed in all cells of the immune system 25 and has been associated with various functional defects of immunosenescence, including failure of immune responses, 33,55 chronic inflammation, 40 and decreased ratio of regulatory:effector T cells. 38 Thus, although immune function in telomerase-deficient mice has been incompletely described as yet, evidence suggests that it could contribute to chronic inflammatory disease in response to enteric bacteria.
Intestinal epithelial abnormalities have been described in telomerase-deficient mice and could contribute to IBD susceptibility, at least in part. In the current study, we described multifocal adenomatous proliferation in the small intestine of TERT and mTR KO mice, as well as proliferation and dysplasia in the colon and cecum. Similar proliferative lesions have been described in telomerase-deficient mice. 21,22 Their pathogenesis is not known, but lack of telomerase is associated with chromosomal instability, particularly in rapidly dividing cell populations, and it is possible that such instability could result in enhanced susceptibility to helicobacter-induced damage and thus predispose to disease. In fact, ulcerative colitis has been associated with short telomeres in humans, 35 although the mechanism is not known. Also, mice that overexpress the TERT component of telomerase have increased resistance to dextran sulfate sodium–induced colitis, supporting the possibility that absence of telomerase activity renders intestinal epithelia more susceptible to injury. 47
In summary, our unexpected finding that telomerase deficiency enhances susceptibility to helicobacter-induced IBD strengthens support for the growing consensus that screening for and elimination of lower bowel helicobacters is necessary to protect genetically engineered mice from disease. 3,9,18 We further showed that even when H hepaticus is not present, other helicobacter species can contribute to naturally occurring disease in mouse colonies and may induce even more severe lesions than the more commonly isolated helicobacters. Finally, we provide preliminary evidence that telomerase deficiency may lead to immunologic or intestinal epithelial dysfunction that could lead to increased susceptibility to disease of the lower bowel.
Footnotes
Acknowledgments
This study was supported in part by Public Health Service grants R01 AI043643 (K.A.E.) and R01 DK070875 (V.B.Y.) from the National Institutes of Health. We thank Paula Arrowsmith and Carrie Schray for outstanding technical assistance with the immunohistochemical staining.
The authors declared that they had no conflicts of interests with respect to their authorship or the publication of this article.
The authors declared that they received no financial support for their research and/or authorship of this article.