
Abstract
The number of Tasmanian devils in the wild is rapidly declining owing to a transmissible cancer, devil facial tumor disease (DFTD). Although progress has been made to understand the spread of this disease, crucial research on the pathogenesis of DFTD has been limited because of the threatened status of the host species. Here, the authors describe the development of a NOD/SCID (nonobese diabetic / severe combined immunodeficiency) mouse model that reproduces DFTD and provides a much-needed model to undertake studies into this intriguing transmissible cancer. Histologically, the DFTD produced in NOD/SCID mice (xenografted DFTD) was indistinguishable from the DFTD identified in Tasmanian devils. At the protein level, all xenografted DFTD tumors expressed periaxin, a marker that confirmed the diagnosis of DFTD. The karyotype of DFTD in NOD/SCID mice reproduced similar chromosomal alterations as seen in diseased devils. Furthermore, each NOD/SCID mouse inoculated with cultured DFTD tumor cells developed tumors, whereas DFTD did not develop in any of the inoculated immune-competent BALB/c mice.
The Tasmanian devil (Sarcophilus harrisii), a marsupial carnivore endemic to Tasmania, Australia, is facing extinction owing to an emergent transmissible cancer, devil facial tumor disease (DFTD). This disease is clearly an important problem, and devils have been recently upgraded to endangered under the Tasmanian and Commonwealth of Australia listings. Without a cure or a vaccine against DFTD, devils may become extinct in the wild within the next 25 to 35 years. 15
It has been proposed that this tumor is transmitted as an allograft from devil to devil during intraspecies facial biting. The tumor-karyotypic arrangement is consistent among affected devils regardless of region, stage of disease, or sex. 20 In addition, genotypic analyses have confirmed that all DFTD tumors are identical clones of transformed cells, in contrast to the genotypes of each affected devil. 17,22 DFTD tumor cell transmission experiments in Tasmanian devils have not formally been reported in the literature. However, unpublished evidence suggests that DFTD can successfully be reproduced in previously unaffected devils after tumor cell inoculation. 21
The mechanism of acceptance of these neoplastic cells by a new host is not yet well understood, primarily as a consequence of the many ethical and financial impediments for undertaking a study of DFTD in diseased devils. In the presence of a competent immune system, 12,13 restriction of major histocompatibility complex genes has been proposed as the main factor in the allograft acceptance of DFTD cells. Experiments with skin grafts among unrelated individuals, however, still need to be performed to prove this hypothesis. 22
With only a limited knowledge of the pathobiology of DFTD, an alternative model is urgently required. NOD/SCID (nonobese diabetic / severe combined immunodeficiency) mice are immunocompromised and provide an ideal alternative model to undertake vital biological studies on DFTD. This model has also been effectively applied to research another naturally occurring transmissible tumor, the canine transmissible venereal tumor (CTVT) in dogs. 7,9 Many aspects of research on the immune response against DFTD would be facilitated through the use of such a laboratory model. Immunization protocols against DFTD, imaging analyses, and incubation period are likely to have direct beneficial applications to understanding the pathobiology of DFTD in Tasmanian devils.
Material and Methods
Establishment of DFTD Tumor Cell Culture
A portion of a primary DFTD tumor (approximately 1 × 1 × 1 cm) was removed from an affected Tasmanian devil. It was rinsed several times with sterile phosphate buffered saline (PBS) supplemented with penicillin (200 U/ml), streptomycin (200 μg/ml), and amphotericin B (0.5 μg/ml; Gibco, Auckland, New Zealand). The tumor was mechanically disaggregated, washed in PBS supplemented with antibiotic, and centrifuged at 300 g for 10 minutes. The DFTD cancer cells were cultured in complete culture medium, composed of RPMI 1640 (Gibco), 10% fetal calf serum (Gibco), 2mM L–glutamine (Sigma–Aldrich, St Louis, MO), and 40 mg/ml of gentamicin (Pfizer, Bentley, Australia) in a humidified 5% carbon dioxide incubator at 35°C. DFTD tumor cells were visually analyzed for confluence and subcultured or cryopreserved as required. Cells were kept in culture for at least 3 months before cytogenetic analyses. When required for inoculations, tumor cells were harvested from the culture flasks, washed twice in sterile PBS, and resuspended at the required concentration.
DFTD Tumor Cell Inoculation
All mice were obtained from the University of Tasmania Central Animal House, housed on a light:dark cycle of 12:12 hours at 21 ± 1°C, and provided with food and water ad libitum. A total of 17 adult NOD/SCID mice (6 weeks old) were injected in the subcutaneous tissue of the right upper flank with viable DFTD tumor cells in a volume of 200 μl of sterile PBS. In addition, 7 NOD/SCID mice were injected with viable cells harvested from masses originated from the previously created xenograft. To investigate whether DFTD could develop from nonviable cells, 7 NOD/SCID mice were injected with nonviable DFTD tumor cells (inactivated by rapid cycles of freezing in liquid nitrogen and thawing in warm water). To further analyze the transmissibility of DFTD, 9 adult BALB/c mice were subcutaneously injected with live cultured DFTD tumor cells. Table 1 summarizes the number of cells injected and the number and strain of mice used. All animal experiments were performed with approval of the Animal Ethics Committee of University of Tasmania (approval No. A10231).
Number of Tumor Cells Inoculated in Mice With the Number and Strain of Mice Used for Inoculations a
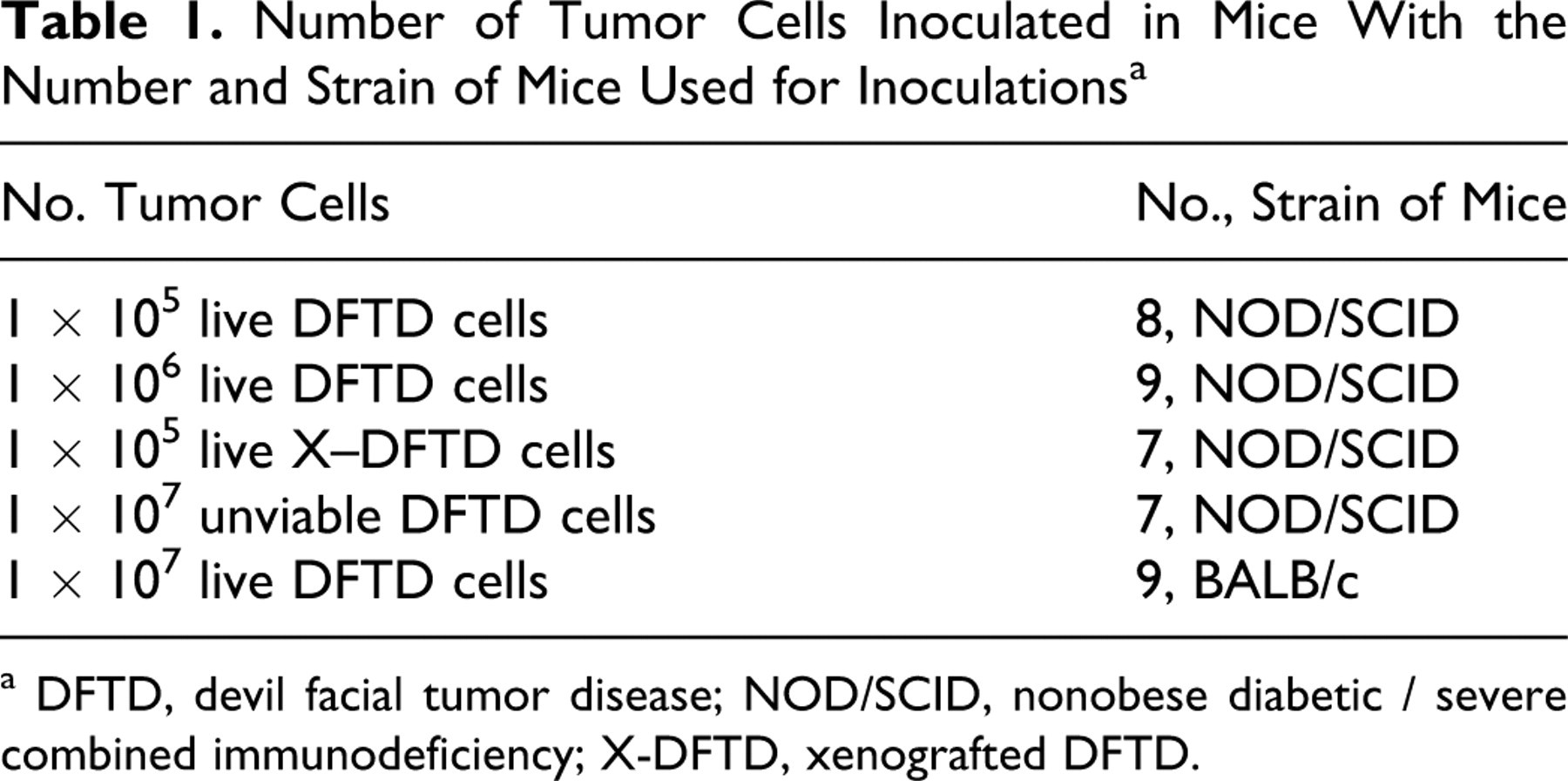
a DFTD, devil facial tumor disease; NOD/SCID, nonobese diabetic / severe combined immunodeficiency; X-DFTD, xenografted DFTD.
Histologic and Immunohistochemical Analyses of Tumor Masses
Mice were euthanized by carbon dioxide asphyxiation when tumor masses were approximately 1 cm3. A thorough necropsy was performed on each mouse, and tissues (whole tumor masses, kidney, lung, heart and liver) were collected and fixed in 10% buffered formalin for at least 24 hours. Collected tissues were processed, embedded in paraffin blocks, and sectioned (3-μm thickness) onto 3-aminotriethoxysilane–coated glass slides. Sections were deparaffinized in xylene, rehydrated through graded alcohol solutions to water, and stained with a standard hematoxylin and eosin (HE) technique.
To confirm that the tumor masses were DFTD and to screen for the presence of DFTD metastasis, tumor masses and organ sections were analyzed for cells expressing periaxin, which is a protein expressed by Schwann cells and which has been proposed as a specific diagnostic marker for DFTD. 17 Briefly, deparaffinized tumor sections were submitted to heat–induced antigen retrieval in citrate buffer (pH, 6; Dako, Carpinteria, CA) with a pressure cooker for 10 minutes and left to cool to 35°C. Endogenous peroxidase activity was quenched by treating the slides in 3% hydrogen peroxide in PBS for 10 minutes at room temperature. The slides were preincubated with blocking serum-free solution (Dako) for 30 minutes and then incubated with rabbit anti–periaxin antibody (1:300, Sigma–Aldrich) for 1 hour. Signal detection was performed with anti-rabbit Dako EnVision+System/HRP (Dako). Sections were counterstained with hematoxylin for 40 seconds and mounted.
Karyotyping
The normal Tasmanian devil karyotype was compared to the karyotype of cultured tumor cells and xenografted tumor cells. For the normal karyotype, peripheral blood lymphocytes from a healthy female Tasmanian devil were utilized. Peripheral blood was cultured in complete RPMI medium and stimulated with phytohemagglutinin (5 μg/ml, Gibco) for 72 hours, whereas the DFTD tumor cells were cultured without the need for stimulation. The peripheral blood cultures were synchronized with thymidine (30 mg/ml, Amersham Pharmacia Biotech, Rydalmere, Australia) and 2-deoxycytidine (0.21 mg/ml, MP Biomedicals, Seven Hills, Australia) to maximize metaphase yield. Colcemid (final concentration, 0.005 μg/ml; Gibco) was added 30 minutes before harvest. The cultured cells were then treated with 0.075M potassium chloride and Carnoy’s fixative (3:1, methanol:glacial acetic acid). Slides were prepared with standard cytogenetic techniques and G banded with trypsin and Leishman stain. 2
Tumor masses harvested from the NOD/SCID mice were cultured in complete RPMI medium. Both tumor cell cultures (DFTD and xenografted DFTD [X–DFTD]) were assessed for confluence; then, Colcemid (final concentration, 0.002 μg/ml) was added for 4 hours before harvest. Cells were removed from the flask with 0.05% trypsin and 0.53 mM EDTA and fixed, with slides being prepared for G banding for karyotypic analysis as described above.
Results
Tumor Development
All NOD/SCID mice inoculated with either 1 × 105 or 1 × 106 viable DFTD tumor cells developed tumors between 4 and 17 weeks after inoculation. Of the mice inoculated with 1 × 105 cells, tumors first became grossly visible at injection sites 7 weeks after the inoculation, and all mice (8 of 8) had visible tumors by 17 weeks. Mice injected with 1 × 106 cells had noticeable subcutaneous tumors 4 to 12 weeks after inoculation. To determine whether DFTD tumor cells transplanted into NOD/SCID mice could reestablish tumor growth, X–DFTD tumors were removed from the mice, and a suspension of single cells was prepared and reinoculated into NOD/SCID mice. All 9 mice inoculated with the second passage of xenografted cells developed tumors by 15 weeks, with the first tumors visible at 5 weeks. Figure 1 illustrates the number of weeks after inoculation in which tumors became first visible. Figure 2 shows the macroscopic appearance of the xenografted tumors in mice.
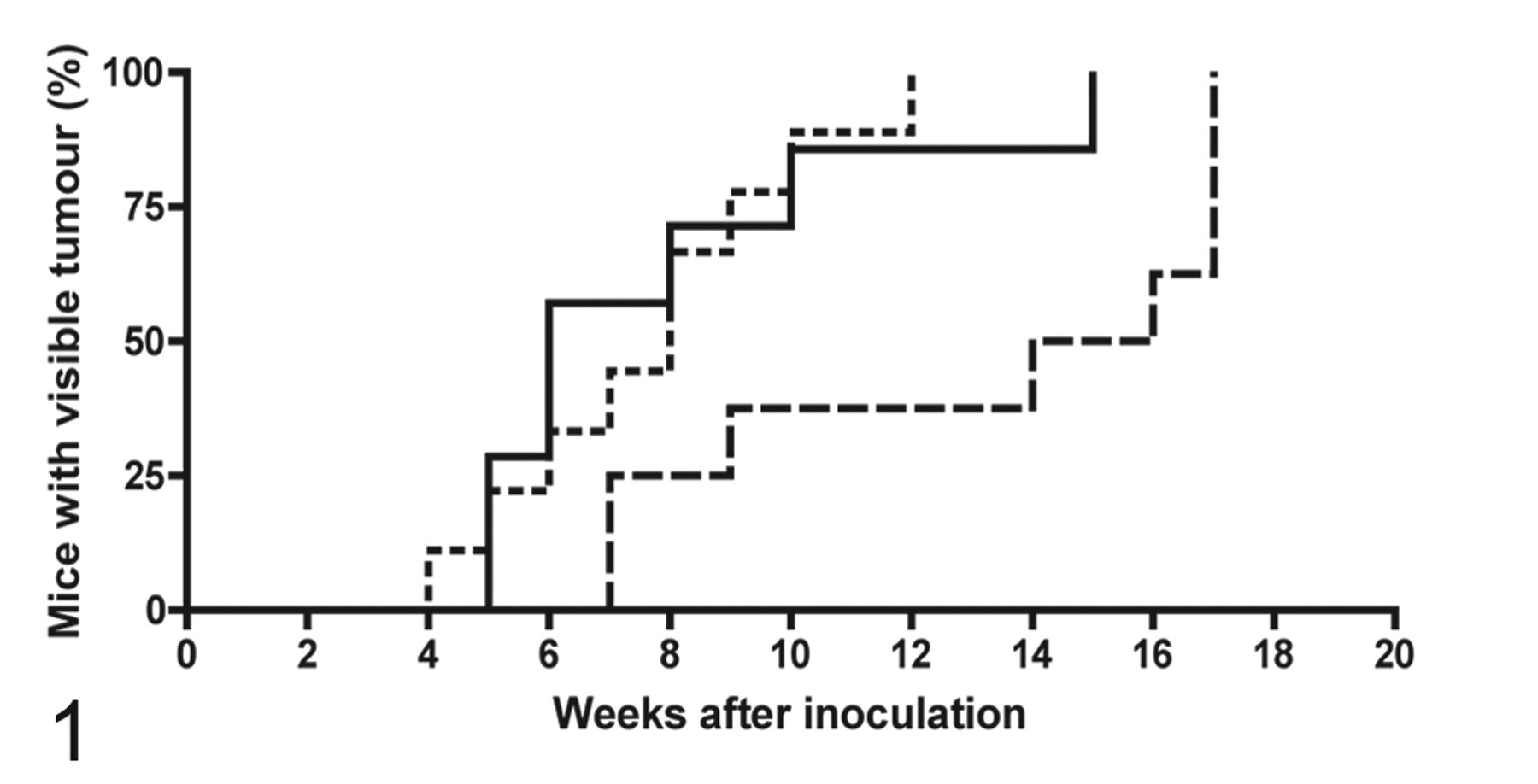
Number of weeks after tumor cell inoculation in which tumors became visible. NOD/SCID (nonobese diabetic / severe combined immunodeficiency) mice inoculated with 1 × 105 devil facial tumor disease (DFTD) cells (long–dash line) had visible tumors between 7 and 17 weeks after inoculation, whereas the same strain of mice inoculated with 1 × 106 DFTD cells (solid line) developed tumors between 5 and 15 weeks after inoculation. NOD/SCID mice inoculated with 1 × 105 xenografted DFTD cells had tumors between 4 and 12 weeks after inoculation (short–dash line).
Mice inoculated with freeze–thaw-killed DFTD tumor cells did not develop tumors for the period of the experiment (30 weeks). Inoculation of live DFTD cells into BALB/c mice did not produce any visible tumors within this 30-week period. Both cohorts were euthanized and necropsied 30 weeks after inoculations, but no evidence of DFTD tumors was detected (data not shown).
Histology and Immunohistochemistry
At necropsy, irregular nodular masses consistent with DFTD tumors were found within the subcutaneous connective tissue layer at the injection sites of the mice inoculated with live DFTD cells. Light microscopic features of the established tumor cells were identical to those seen in primary DFTD tumors in Tasmanian devils. No metastases were found in any of the affected mice.
Viable tumor cells were polyhedral or ovoid with an acentric nuclei. Tumor cells appeared in situ as loosely nested aggregates or as lines of cells parallel to the connective tissue stroma. On close examination, a high proportion of tumor cells were in mitosis, with up to 5 to 7 tumor cells in high-power fields (400×) displaying prominently stained metaphase/telophase chromosomes.
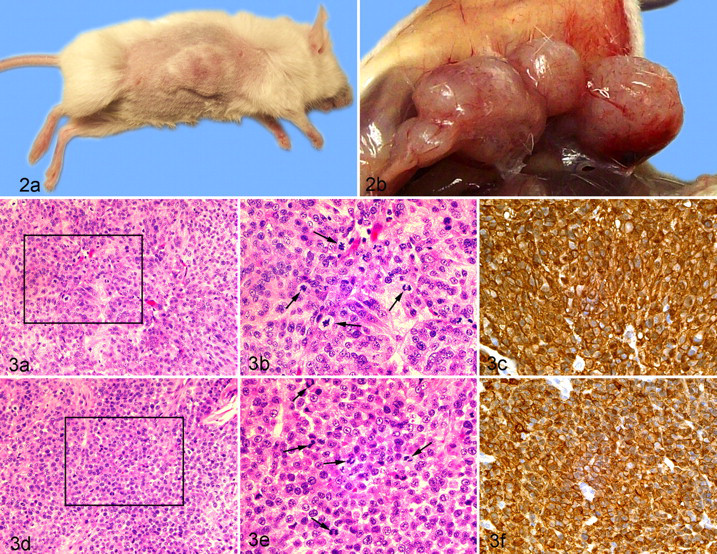
The blood vessels directly sustaining these transplanted tumors were particularly prominent at necropsy. At the histologic level, the microvascularization within growing tumor aggregates consisted of interspersed capillaries supported by minimal connective tissue stromal elements. Tumors cells were loosely clumped with no evidence of recognized intercellular junctions between cells. The tumor growth at the injection nidus appeared to be a passive process rather than one characterized by any active invasion of surrounding normal tissue by tumor cells. The localized expansion of the growing tumors was assisted by the apparent ease of separation of individual tumor cells from a proliferating aggregate. Over time, apoptosis and generalized avascular necrosis were evident in growing tumor masses. Sections of X–DFTD labeled strongly with periaxin, indicating that the passaged tumor maintained its antigenic attribute. Figure 3 shows HE- and periaxin-labeled sections of a X–DFTD tumor in a mouse (Fig. 3A–3C) and a primary DFTD tumor (3D–3F) in a Tasmanian devil.
Cytogenetics
A standard devil karyotype was established as a reference to clarify the cytogenetic changes associated with DFTD and X–DFTD tumor cells. There is no cytogenetic ideogram available for Tasmanian devils; therefore, the described karyotypes have been extrapolated with human-cytogenetic first principles. The normal karyotype of the Tasmanian devil consists of 14 chromosomes: 12 autosomes and XY and XX sex chromosomes in males and females, respectively.
The DFTD tumor karyotype was consistent with that published by Pearse and Swift in 2006, 20 with minor modifications based on our interpretation of the chromosomes. These modifications included the realignment of a chromosome 5 as an abnormal chromosome 6 and the identification of one marker chromosome as a deleted chromosome 5. The most commonly observed DFTD karyotype (64% of analyzed cells) was a grossly abnormal pseudodiploid karyotype with a total of 14 chromosomes. Loss of the sex chromosomes and both chromosome 2 homologues was observed in addition to deletions of the long arm of chromosome 1 and the entire long arm of chromosome 5. There was additional material on the short arm of chromosome 6 as well as four unidentifiable marker chromosomes. Two different subclones were also observed, showing evidence of clonal variation. These two subclones consisted of a near-triploid karyotype (approximately 21% of cells) and a near tetraploid with additional structural and numerical abnormalities (approximately 15% of cells).
The X–DFTD karyotype displayed the same chromosomal features of the pseudodiploid karyotype of the DFTD subclone. Approximately 39% of cells recovered from NOD/SCID mice retained an identical karyotype to the most common DFTD cell clone, with a modal number of 14. These included the loss of the sex chromosomes, a deletion of the long arm of chromosome 1, the loss of both chromosome 2 homologues, abnormalities of chromosomes 5 and 6, and four unidentifiable marker chromosomes. Many of the observed cells reflected the triploid (46%) and near-tetraploid (15%) DFTD karyotypes displaying similar structural and numerical changes. Table 2 summarizes the composite karyotype and number of cells analyzed for the DFTD and X–DFTD tumor cells. Figure 4 shows the constitutional devil karyotype, the most common DFTD and X–DFTD karyotypes, and a triploid to tetraploid karyotype.
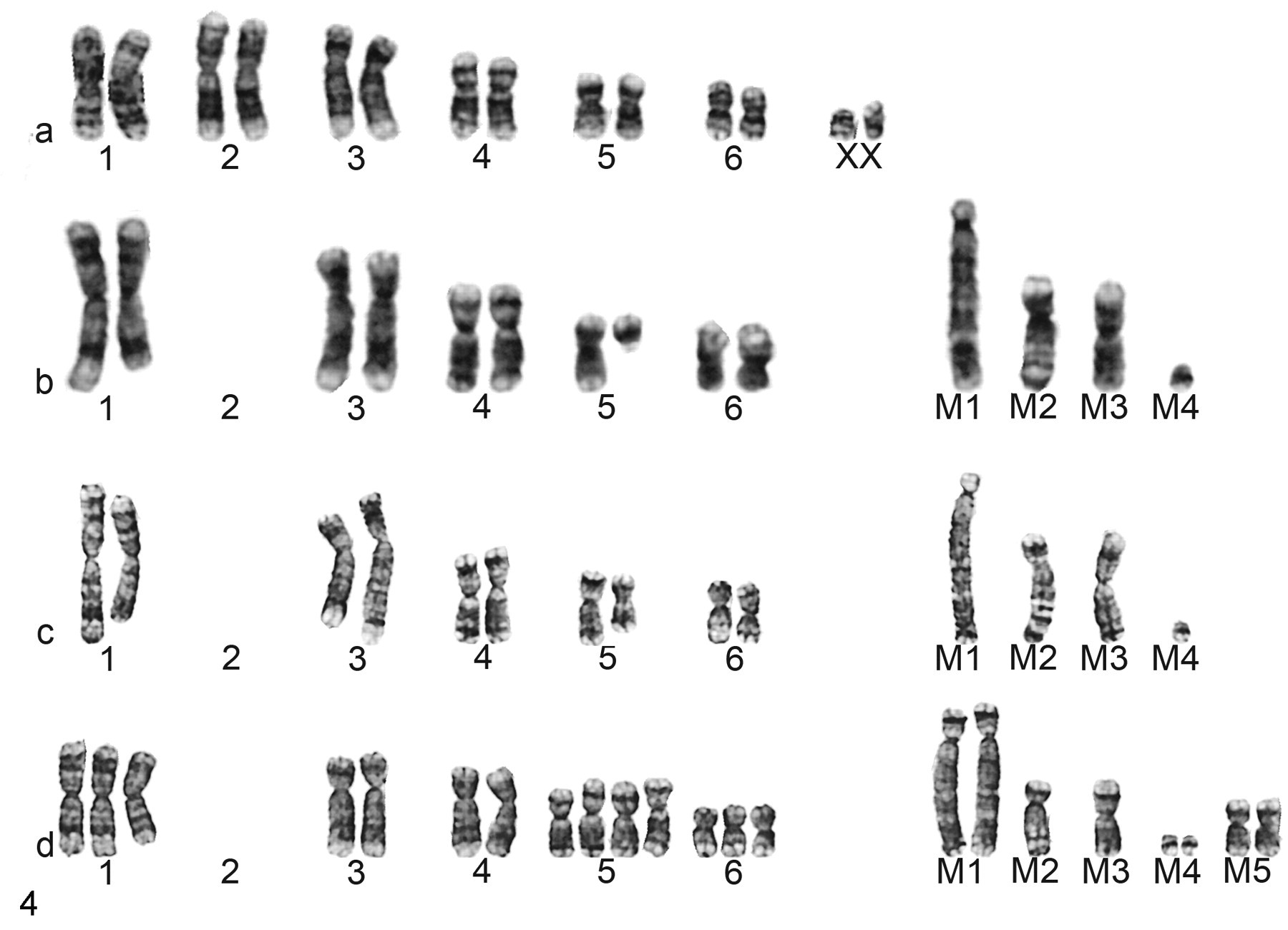
Karyotype analyses of constitutional and devil facial tumor disease–derived tumor cells. Constitutional female Tasmanian devil cell with 12 autosomes and 2 XX sex chromosomes (A). The karyotypic arrangement of devil facial tumor disease tumor cell chromosomes contains aberrant alterations, such as the loss of both chromosomes 2 and the sex chromosomes and 4 additional markers (M1–M4) (B). These alterations were preserved after the mouse passage (C). Occasional triploid and near-tetraploid clones were observed in the primary devil facial tumor disease culture (not shown); these clones were abundant in the mouse karyotype (D).
Composite Karyotype and Number of DFTD and X–DFTD Cells Analyzed a

a DFTD, devil facial tumor disease; X-DFTD, xenografted DFTD. Note that the karyotypes are composite karyotypes, indicating that not all cells analyzed contained all the described abnormalities. Numbers in parentheses represent the number of karyotypes observed out of total number counted.
Discussion
Neoplasia is generally not thought of as a disease process associated with social interactions, unless an exogenous agent (such as a virus) is directly implicated in tumorigenesis and infectious transmission. Thus, DFTD is somewhat unique because it is a socially transmissible tumor disease, transferred as an allograft in a wild-species population. In this aspect, it resembles CTVT in dogs, which is also naturally transferrable by social interactions, particularly by venereal transmission. Although the mechanisms of CTVT tumor acceptance have been more extensively examined, 10,11,18 little is known about the host–agent immunobiology of DFTD, partially because of inaccessibility to DFTD–affected devils.
In our murine model, viable DFTD tumor cells injected into immunocompromised NOD/SCID mice successfully established the tumor in all mice. Tumor cells derived from these masses could be passaged into other NOD/SCID mice, thereby demonstrating that the tumor cells grown in laboratory mice mimicked the transmissibility of DFTD. The diagnostic characteristics of DFTD, such as its distinctive chromosomal abnormalities, histologic features, and periaxin expression, were preserved after the xenotransplantation and passage in mice. This is encouraging for the scope of potential DFTD research using laboratory-maintained NOD/SCID mice.
Unlike wild DFTD–affected devils, which have an incidence of 65% of metastases, 14 no mouse was identified bearing metastasis related to X–DFTD development. The requirement to euthanize NOD/SCID mice bearing tumor masses that had achieved 1 cm3 in volume, for animal welfare reasons, may have reduced the likelihood of detecting DFTD metastases in these mice.
The interval between inoculation and the first physical evidence of in situ tumor growth in mice varied. Although the tumor cell viability after inoculation was not measured, it is likely that many tumor cells died immediately following the subcutaneous inoculation. Consequently, the number of surviving inoculated cells would have varied among different mice. It has been observed that approximately 10% of injected CTVT tumor cells remained viable 7 days after iatrogenic inoculation in dogs and immunocompromised mice, 9 which suggests that only a small population of viable cells was required to establish a tumor mass.
Macroscopically, X–DFTD was similar to DFTD from wild devils and microscopically consistent with the previously described primary DFTD tumors. 14 The key features of DFTD, which include the polyhedral to ovoid nature of the cells in loosely nested aggregates, prominent blood vessels, and lack of lymphocyte infiltration, were faithfully reproduced with X–DFTD. The lack of lymphocyte infiltration would be expected with this model, given that NOD/SCID mice have impaired adaptive immune functions (T– and B–cell competency) and defects in innate immunity (natural killer cell activity). 6 Because X–DFTD failed to develop in normal immune-competent BALB/c mice, there is evidence that the immune response can prevent the establishment of DFTD, thereby providing an avenue through which to investigate the immune response to DFTD.
Rapid cell multiplication was evident by the high number of mitotic figures present, leading to the rapid expansion in tumor mass. The growth rate and incubation period of DFTD in devils are unknown. It is likely, however, that our mouse model could be useful in determining optimal tumor cell growth rates by comparison of mitosis figures seen microscopically among these tumors in mice and devils. The absence of intercellular junctions would allow the tumor cells at the periphery of growing tumor masses to be subject to effortless spread and colonization of the loose connective tissues. Consequently, it is likely that metastases would have eventually developed similar to the primary tumors of Tasmanian devils.
DFTD tumor cells express a range of myelination proteins, such as periaxin, peripheral myelin protein 22, and nerve growth factor receptor. Based on this, a recent proposal posited that DFTD is derived from Schwann cells from the peripheral nerve system. 17 High expression of periaxin was confirmed in the xenografted tumors, clearly demonstrating that these masses were DFTD cells. It would appear that the mouse infection with DFTD tumor cells did not alter greatly the protein profile of these cells.
Cytogenetically, the transplanted X–DFTD tumor cells retained the chromosomal abnormalities evident in the primary DFTD tumor. Many of the subclones identified in the DFTD cell line were also detected in the X–DFTD cell line. There was a possible proliferative advantage of the triploid cell population, as evidenced by the detection of a larger population of those cells. These X–DFTD cells were also identified in culture before inoculation, and it is likely that this cell population developed as relatively stable clones in vitro and was maintained following inoculation.
The DFTD karyotype has abundant structural and numerical alterations, and in the absence of more elaborate techniques, such as fluorescent in situ hybridization or chromosome painting, it was difficult to match the chromosomes. Consequently, with some karyotypes approaching triploidy/tetraploidy, alignment was difficult and not necessarily precise. Despite this, the karyotypes clearly show the features of DFTD chromosomes and highlight the substantial chromosomal alterations characteristic of this tumor.
To investigate whether an exogenous cause, independent of the live transformed cells, could be incriminated in the development of DFTD, NOD/SCID mice were inoculated with nonviable DFTD tumor cells. None of these mice produced any tumor masses, which is consistent with the proposal that DFTD occurs following transmission of viable cell transplants rather than as transmission of an exogenous pathogen acting as the causal agent.
The inability of DFTD to develop in immunocompetent BALB/c mice suggests that the potential for natural transmission of DFTD to other mammalian hosts with competent immune systems is unlikely. This supports field observations that DFTD is a species–specific neoplasm. 8 Further transmission trial experiments, using animals more closely related to the devil (eg, the marsupial model Monodelphis domestica), would be the required step to investigate whether DFTD tumor cells can infect other species.
Immunocompromised mice with human–reconstituted immune systems have been used for the past two decades to study immune responses in an in vivo model. 4,16,19 This model has been adapted to other species in which such studies are difficult to perform in a controlled environment, such as large farm animals and cetaceans. 1,3,5 In the case of the Tasmanian devil, the numbers allowed for such research are limited owing to the threatened status of the species. The description of a murine model for DFTD was the first step toward the development of a devil–reconstituted NOD/SCID mouse model, which may be useful to investigate immune responses and the development of a vaccine against DFTD.
Footnotes
Acknowledgements
We are grateful to the staff from the Central Animal House of University of Tasmania for help with the mice and to Ms Narelle Phillips for invaluable help with histologic preparations. Financial support was provided by a Dr Eric Guiler Tasmanian Devil Research Grant, and A.K. was supported by an Australian Research Council linkage grant.